10-K: Annual report pursuant to Section 13 and 15(d)
Published on October 15, 2018
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K
(Mark one)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 or 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended July 31, 2018
or
| o | TRANSITION REPORT PURSUANT TO SECTION 13 or 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ______________ to ______________
Commission File Number 001-09974
| ENZO BIOCHEM, INC. | ||
| (Exact name of registrant as specified in its charter) |
| New York | 13-2866202 | |
| (State or other jurisdiction | (I.R.S. Employer | |
| of incorporation or organization) | Identification No.) | |
| 527 Madison Ave. | ||
| New York, New York | 10022 | |
| (Address of principal executive offices) | (Zip Code) |
| (212) 583-0100 | ||
| (Registrant’s telephone number, including area code) |
Securities registered pursuant to Section 12(b) of the Act:
| (Title of Each Class) | (Name of Each Exchange on Which Registered) | |
| Common Stock, $.01 par value | The New York Stock Exchange |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes o No x
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes x No o
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K
Yes x No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer , a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer o |
Accelerated filer x |
Non-accelerated filer o |
Smaller reporting company o |
Emerging growth company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act of 1934). Yes o No x
The aggregate market value of the registrant’s voting stock held by non-affiliates of the registrant was approximately $318,221,000 as of January 31, 2018.
The number of shares of the Company’s common stock, $.01 par value, outstanding at September 30, 2018 was 47,182,254.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive Proxy Statement to be delivered to shareholders in connection with the Annual Meeting of Shareholders to be held on or about January 3, 2019 are incorporated by reference into Part III of this annual report.
TABLE OF CONTENTS
| 1 |
PART I
Overview
Enzo Biochem, Inc. (the “Company” “we”, “our” or “Enzo”) is an integrated diagnostic bioscience company focusing on delivering and applying advanced technology capabilities to produce affordable reliable products and services to allow our customers to meet their clinical needs. We develop, manufacture and sell our proprietary technology solutions and platforms to clinical laboratories, specialty clinics and researchers and physicians globally. Enzo’s structure and business strategy represent the culmination of years of extensive planning and work. The Company now has the unique ability to offer low cost, high performance products and services in molecular diagnostics, which ideally positions us to capitalize on the reimbursement pressures facing diagnostic labs. Our pioneering work in genomic analysis coupled with our extensive patent estate and enabling platforms have positioned the Company to continue to play an important role in the rapidly growing molecular medicine marketplaces.
Enzo technology solutions and platforms and unique operational structure are designed to reduce overall healthcare costs for both government and private insurers. Our proprietary technology platforms reduces our customers' need for multiple, specialized instruments, and offer a variety of high throughput capabilities together with a demonstrated high level of accuracy and reproducibility. Our genetic test panels are focused on large and growing markets primarily in the areas of personalized medicine, women's health, infectious diseases and genetic disorders.
For example, our AMPIPROBE® technology platform can lead to the development of an entire line of nucleic acid clinical products that can allow laboratories to offer a complete menu of services at a cost that allows them to enjoy an acceptable margin. Our technology solutions provide tools to physicians, clinicians and other healthcare providers to improve detection, treatment and monitoring of a broad spectrum of diseases and conditions. In addition, reduced patient to physician office visits translates into lower healthcare processing costs and greater patient services.
In the course of our research and development activities, we have built a substantial portfolio of intellectual property assets, comprised of 343 issued patents worldwide and over 157 pending patent applications, along with extensive enabling technologies and platforms.
Operating Segments
We are comprised of three interconnected operating segments which have evolved out of our core competencies involving the use of nucleic acids as informational molecules and the use of compounds for immune modulation and augmented by the previous acquisitions of a number of related companies. Information concerning sales by geographic area and business segments for the years ended July 31, 2018, 2017 and 2016 is located in Note 15 in the Notes to Consolidated Financial Statements.
Below are brief descriptions of each of our operating segments:
Enzo Clinical Laboratory Services is a clinical reference laboratory providing a wide range of clinical services to physicians, medical centers, other clinical labs and pharmaceutical companies. The Company believes having a CLIA-certified and a College of American Pathologists (“CAP”) accredited medical laboratory located in New York provides us the opportunity to more rapidly introduce cutting edge products and services to the clinical marketplace. Enzo Clinical Labs offers an extensive menu of molecular and other clinical laboratory tests and procedures used in patient care by physicians to establish or support a diagnosis, monitor treatment or medication, and search for an otherwise undiagnosed condition. Our laboratory is equipped with state-of-the-art communication and connectivity solutions enabling the rapid transmission, analysis and interpretation of generated data. We operate a full service clinical laboratory in Farmingdale, New York, a network of over 30 patient service centers throughout New York, New Jersey and expanding into Connecticut, a free standing “STAT” or rapid response laboratory in New York City and a full service phlebotomy, in-house logistics department, and an information technology department. Given our license in New York State, we are able to offer testing services to clinical laboratories and physicians in the majority of states nationwide.
Enzo Life Sciences Products manufactures, develops and markets products and tools for clinical research, drug development and bioscience research customers worldwide. Underpinned by broad technological capabilities, Enzo Life Sciences has developed proprietary products used in the identification of genomic information by laboratories around the world. Information regarding our technologies can be found in the “Core Technologies” section. We are internationally recognized and acknowledged as a leader in the development, manufacturing validation and commercialization of numerous products serving not only the clinical research market, but also the life sciences markets in the fields of cellular analysis and drug discovery, among others. Our operations are supported by global operations allowing for the efficient marketing and delivery of our products around the world.
| 2 |
Enzo Therapeutics is a biopharmaceutical venture that has developed multiple novel approaches in the areas of gastrointestinal, infectious, ophthalmic and metabolic diseases, many of which are derived from the pioneering work of Enzo Life Sciences. Enzo Therapeutics has focused its efforts on developing treatment regimens for diseases and conditions for which current treatment options are ineffective, costly, and/or cause unwanted side effects. This focus has generated a clinical and preclinical pipeline, as well as more than 154 patents and patent applications.
The Company’s primary sources of revenue have historically been from the clinical laboratory services provided to the healthcare community and product revenues, royalty and licensing of Enzo Life Sciences’ products utilized by customers worldwide. The following table summarizes the sources of revenues for the fiscal years ended July 31, 2018, 2017 and 2016 (in thousands and percentages):
| Fiscal year ended July 31, | 2018 | 2017 | 2016 | ||||||||||||||||||||
| Clinical laboratory services | $ | 74,777 | 71 | % | $ | 77,407 | 72 | % | $ | 70,915 | 69 | % | |||||||||||
| Product revenues | 29,224 | 28 | 29,192 | 27 | 30,337 | 30 | |||||||||||||||||
| Royalty and license fee income | 712 | 1 | 1,205 | 1 | 1,521 | 1 | |||||||||||||||||
| Total | $ | 104,713 | 100 | % | $ | 107,804 | 100 | % | $ | 102,773 | 100 | % | |||||||||||
Markets
Clinical diagnostics
The U.S. clinical diagnostics market has been reported by industry sources to be greater than $25 billion annually and over $60 billion worldwide. It is comprised of a broad range of tests based on clinical chemistry, microbiology, immunoassays, genomics, proteomics, gene expression profiling, blood banking, and cancer screening assays through histology as well as newer body fluid based approaches. Many of these tests employ traditional technologies such as cell culture technologies.
Immunoassays are based on the use of antibodies directed against a specific target or antigen to detect that antigen in a patient sample. Cell culturing techniques involve the growth, isolation and visual detection of the presence of a microorganism and often its susceptibility to FDA approved drugs.
There are several drawbacks to these more traditional technologies. Immunoassays do not allow for early detection of diseases because they require minimum levels of antigens to be produced by the microorganism in order to be identified. These levels vary by microorganism, and the delay involved could be several days or several months, as seen in HIV/AIDS. Cell cultures are slow, labor intensive and not amenable to all microorganisms. For example, gonorrhoea and chlamydia are difficult to culture.
Molecular diagnostics have many advantages over traditional technologies. Since gene-based diagnostics focus on the identification of diseases at the molecular level, they can identify the presence of the disease at its earliest stage of manifestation in the body. These tests provide results more rapidly, are applicable to a broad spectrum of microorganisms and can easily be automated in a multiplex platform.
Several advances in technology are accelerating the adoption of gene-based diagnostics in clinical laboratories. These advances include high throughput automated formats that minimize labor costs, non-radioactive probes and reagents that are safe to handle, and amplification technologies that improve the sensitivity of such diagnostics.
According to industry sources, the market for molecular diagnostic tools, assays and other products is currently more than $7 billion per year, and is acknowledged as one of the fastest growing segments in the in-vitro diagnostic industry, growing at more than twice the rate of traditional diagnostics. Contributing to this growth are, among other factors:
| • | the increasing number of diagnostic tests being developed from discoveries in genome research; |
| • | advances in formats and other technologies that automate and accelerate gene-based diagnostic testing; |
| • | growing emphasis by the healthcare industry on early diagnosis and treatment of disease and; |
| • | application of gene-based diagnostics as tools to match therapies to specific patient genetics, commonly referred to as pharmacogenomics or companion diagnostics. |
| 3 |
Diagnostic Products
There is a large and growing global demand by biomedical and pharmaceutical companies for research and diagnostic tools that both facilitate and accelerate the generation of biological information. This demand can be met by gene and protein target-based diagnostics for which a variety of formats and tools have been developed that enable researchers to study biological pathways. These tools can identify mutations in gene sequences and variations in gene expression levels that can lead to disease, or they can quantify biomarkers that provide insight into disease and potential therapeutic solutions. These techniques use instruments such as DNA sequencing and genotyping equipment, microarrays, fluorescent microscopes, high content screening platforms, flow cytometers and plate readers. Common among these instruments is the need for reagents that allow the identification, quantification and characterization of interactions of specific genes or nucleic acid sequences, proteins, cells, and other cellular structures and organelles.
We believe this market will continue to grow as a result of:
| • | long term commitment to research spending by academic, government and private organizations to determine the function and clinical relevance of the gene sequences and proteins that have been identified by genomics research, |
| • | development of commercial applications based on information derived from this research and, |
| • | on-going advancements in tools that accelerate these research and development activities. |
Therapeutics
We believe our core technologies have broad diagnostic and therapeutic applications. We have focused our efforts on discovering how best to treat pathologies associated with bone or metabolic control and immune-mediated diseases. Although the causes of disorders such as Crohn’s disease, autoimmune uveitis and non-alcoholic steatohepatitis (NASH) remain unknown, various features suggest immune system involvement in their pathogenesis.
We continue to test technologies we believe can serve as enabling platforms for developing medicines that genetically target and inhibit viral functions, as well as medicines that regulate the immune response. In addition to such therapeutic products, we continue to capitalize on our nucleic acid labeling, target and signal amplification, and detection technologies and intellectual property to develop diagnostic and monitoring tests for various diseases.
We believe our expertise in developing and securing approvals of novel platform technologies will enable us to shorten the development time and capture meaningful market share.
Strategy
Our objective is to develop and manufacture high value affordable and reliable diagnostic products and services using our proprietary technologies to allow our customers to meet their clinical needs. Our proprietary technology platforms, if successful, will alter the existing business models and improve economics across the healthcare industry. Our strong intellectual property estate provides freedom to operate and compete in a rapidly growing molecular diagnostic healthcare marketplace.
We believe our expertise in developing and marketing proprietary technology platforms uniquely positions Enzo to provide products and services that will change the fundamental relationship between molecular diagnostic companies and clinical laboratories. Our technology platforms will provide economic and market optionality to use Enzo’s products and services for margin improvement. As such, clinical laboratories will be able to enter and compete in markets that until now have been out of reach due to poor economic standing caused by high costs of reagents and equipment rental arrangements from molecular diagnostic companies coupled with lower reimbursement from governmental and commercial healthcare companies.
Our objective allows clinical laboratories to purchase low cost reagents and kits to be run on open system platforms already in use in their labs, or to use Enzo as a low cost reference laboratory. Enzo’s integrated business model not only provides benefits to clinical laboratories, but also to insurance providers who will benefit from more clinical laboratories able to compete for testing services with national laboratories.
In addition to selling these highly effective and compatible platforms and their assays, we are positioning ourselves as a reference lab for independent labs nationwide primarily by offering lower cost reference services.
Our commitment to utilizing our proprietary technologies to develop clinically relevant diagnostics, while helping to relieve the cost pressures that independent laboratories are bearing is core to our strategy. It underscores the progress we are achieving in our strategy
| 4 |
of utilizing Enzo’s integrated structure to produce diagnostic products and services relevant to today’s dynamic and challenging healthcare marketplace.
By developing a broad technology base, Enzo has positioned the Company for a robust flow of products and services that will provide medically relevant, cost effective solutions easily adaptable to the workflow of the clinical laboratory, and its ability to do so is based on several factors, including:
| · | The Company’s integrated structure that enables it to internally develop and advance products seamlessly from innovation through commercialization validation via recent patent settlements of Enzo’s intellectual property strength and ownership of basic patents that provide an economic advantage. |
| · | The unique ability to deliver high performance, easily adaptable products and services that are also cost effective for independent labs as well as Enzo’s own clinical lab in a steadily declining reimbursement environment |
| · | Ample finances with which to execute and follow through on the Company’s integrated strategy. |
Increase investment in research and development & product development
We are increasing our research and development efforts to develop new leading edge solutions in the rapidly growing molecular diagnostic marketplace. Current technology platforms under development include:
| · | AMPIPROBE® Platform – easily adaptable, affordable, real time DNA amplification and detection |
| · | FLOWSCRIPT® – enhanced flow cytometry for single cell analysis |
| · | Enhanced Immunohistochemistry – moving Pathology to the next generation |
| · | Enhanced Immunoassays – pushing sensitivity to expand immunoassay applications |
| · | Polyview Plus® Systems- optimized reagents for clear, consistent IHC and ISH results |
Enzo’s proprietary platforms and the related assays developed can provide more sensitive diagnostic information at lower costs than many other tests currently marketed. The Company designs its products to be able to work with lower specimen volume which not only allows the laboratories to run more tests off of a single clinical specimen, but also may reduce the need for patients to submit additional samples, thus reducing unnecessary physician visits. The Company’s newly approved assays are the forerunners of a comprehensive line of diagnostic solutions under development by Enzo to address the critical needs of clinical laboratories that are often locked into closed-system contracts with molecular diagnostics suppliers that, with ever-declining reimbursements, reduce or even eliminate operating margins.
New MDx Platform
Enzo has developed and validated a new molecular diagnostics platform, unlike current closed system platforms, that has an open access feature, flexibility and compatibility with a full clinical workflow. It is believed to be uniquely characterized as an automated, clinically compatible, open platform that operates with multiple reagents and sample types allowing for cost savings and LDT tests (Laboratory Developed Test), while remaining comparable with FDA-approved products. It has been optimized to fully address existing clinical work flows while also providing the flexibility to develop and incorporate new work flows. The clinical diagnostic industry is challenged by declining reimbursements and high reagent costs associated with “closed systems” diagnostic platforms that prohibit the use of third-party reagents. The new Enzo “open system” molecular diagnostic platform is compatible with existing sample collection devices. It runs on a standardized, simplified sample processing (fluid handling and nucleic acid isolation) and amplification/detection workflow, and was designed to provide high performance and adaptable solutions to existing lab workflow, while addressing the critical need for lower cost solutions. The platform is compatible with a sample input capacity up to 1 ml of whole blood, serum, plasma, urine, gynecological and non-gynecological specimens, and offers high throughput, 96-well plate molecular testing in less than four hours run time. At full capacity, the platform can process as many as three 96-well plates (268 samples total) in about eight hours for most of the company’s tests. Enzo has developed multi-target viral load assays and multi-target DNA-based women’s health assays optimized for the new automated, open system platform, and is currently in the process of developing a screening assay for oncogenic forms of HPV. The platform has current compatibility with more than 16 Enzo-developed clinical tests in the areas of sexual health, women’s health, virology, upper respiratory infections, plus others, with a built-in capacity to run new or esoteric laboratory developed tests.
Continue to Commercialize New Platforms for Molecular Diagnostics via Multiple Channels
We have developed several enabling platform technologies that may have utility in the development of a new generation of molecular diagnostic products designed to meet the needs of the current clinical marketplace. Our lead solution is AMPIPROBE® platform, which is proprietary target amplification and detection technology that has been shown to require substantially less starting material than conventional methods such as polymerase chain reaction (PCR) based products. With AMPIPROBE® platform it may be possible to increase the number of analytes that can be assayed for from a single clinical specimen, which in turn may reduce the need for physicians to recall patients to obtain additional clinical material for testing. In addition by increasing the number of analytes tested in a single clinical preparation, AMPIPROBE® platform may be able to produce diagnostic tests at a significantly lower cost than conventional assays. Moreover, the need for less starting material may also lead to diagnostic tests with improved sensitivity, thus allowing detection of certain analytes present in minute quantities that are below the limit of detection of conventional assays.
We have already introduced the first product using our FLOWSCRIPT® platform technology for the identification of gene expression in clinical samples specifically detection of mRNA from Human papillomavirus (HPV) oncogenes E6 and E7. Overexpression of these HPV oncogenes promotes the growth of malignant cells, leading to the development of cervical cancer. The FLOWSCRIPT® technology platform is a proprietary, flow cytometry-based molecular detection system for the multiplex analysis of cell function and identity and was developed by cross-functional teams at Enzo. The HPV E6/E7 assay is the first product to utilize this novel platform. Analysis is performed on a small volume of a liquid cytology specimen and can thus be easily incorporated as a reflex test measure following abnormal Pap smear results. The assay, and the platform on which it is based, allows for the simultaneous analysis of several different genes expressed in every cell in a given sample. In this manner, it is possible to produce clinically relevant data at the single cell level. Unlike other assays that study mRNA expression, FLOWSCRIPT® assays are performed using a homogeneous system that eliminates washing steps that can reduce fluctuation of results. Additionally, the assay’s use of external control improves run-to-run consistency. As a result, both hands on time and the number of steps are reduced, allowing for improved economics. In data presented at a 2015 pathology conference in Italy, Enzo’s assay was shown to produce reliable and consistent results near the limit of assay detection. Furthermore, Enzo anticipates using this platform for a multitude of applications such as study of other cancers and the evaluation of an individual immune state as well as products targeted to the drug development market, among others.
| 5 |
The FLOWSCRIPT® platform is used to help guide providers in assessing the risk of progression to cervical cancer and whether
colposcopy or follow-up screening should be the preferred course of action. This assay demonstrates Enzo’s commitment to
utilizing our proprietary technology and bringing forward clinically relevant diagnostics that can inform patient and physician
decision-making with potential to reduce spending associated with advanced stage disease. Moreover, it is indicative of how well
we are executing our strategy of utilizing our integrated structure to produce products that are relevant to today’s evolving
healthcare marketplace.
Maximize our resources by collaborating with others in therapeutics research and commercialization activities
We enter into research collaborations with leading academic and other research centers to augment our core expertise on specific programs.
Our clinical trial of OPTIQUEL® is a direct result of a research collaboration; we acquired the rights and intellectual property to this candidate drug and technology intended for use in the treatment of autoimmune uveitis. Working with scientists and physicians in the United States and abroad, Enzo continued drug development to the stage of a clinical trial now in further evaluation by the National Eye Institute of the National Institutes of Health in Washington DC.
We have research and clinical collaborations with other institutions including Hadassah University Medical Center in Jerusalem, Israel relating to our immune regulation technology. Through collaborations such as these and other licensing agreements we continue to develop novel therapeutics for the stimulation and enhancement of bone formation and glucose control, among others. Products emanating from this technology could provide potential therapy for bone disorders, including bone loss, bone fractures, periodontitis, diabetes and other indications. There can be no assurance that any of these collaborative projects will be successful.
Similarly, we may seek to fully exploit the commercial value of our technology by partnering with for-profit enterprises in specific areas in order to act on opportunities that can be accretive to our efforts in accelerating our development program.
Exploit our marketing and distribution infrastructure
Enzo Life Sciences maintains relationships with academic and commercial groups worldwide in sourcing and commercializing high value reagents developed by leading academics. We have also developed a sales and marketing infrastructure to directly service its end users such as clinical laboratories, researchers and pharmaceutical companies, while simultaneously positioning the Company for targeted product line expansion. Our global sales, marketing, manufacturing, product development and distribution infrastructure, have now been integrated and consolidated into a single global business. Enzo Life Sciences operates, under its own name, worldwide through wholly owned subsidiaries (in USA, Switzerland, Benelux, Germany, and the UK), a branch office in France and a network of third party distributors in most other significant markets worldwide. Our comprehensive product portfolio allows us to deliver integrated solutions to basic researchers, drug developers and clinical researchers around the globe. Our research allows us to provide solutions in all key research areas including: Genomics, Cell Biology, Immunoassays, and in a multitude of applied research markets including: Bioprocess, Personal Care, Cancer Research, and Neuroscience to name a few.
Expand and protect our intellectual property estate
Since our inception, we have followed a strategy of creating a broad encompassing patent position in the life sciences and therapeutics areas. We have made obtaining patent protection a central strategic policy, both with respect to our proprietary platform technologies and products, as well as broadly in the areas of our research activities. During fiscal 2018, we were issued 40 patents and expanded our patent estate in the area of nucleotides, amplification, labeling and detection, among others.
Product Development and Pipeline
Enzo is committed to delivering a robust line of products and services
that will provide medically relevant, cost effective solutions that are easily adaptable to the workflow of clinical laboratories. The
Company’s integrated Life Sciences-Clinical Lab structure continues to be instrumental in its ability to seamlessly develop
and advance products from innovation and manufacturing in our life sciences group to validation and commercialization through our
clinical laboratory.
The Company’s development pipeline includes an extensive line of assays for detection of numerous women’s health infectious agents as well as for the identification of other pathogens. The Company is also developing a proprietary line of products designed to aid pathologists in differentiating the characteristics of various tumors from biopsy specimens. The Company’s molecular products and services are targeted at a market currently estimated to be in excess of $10 billion annually.
| 6 |
During fiscal 2016 and more recently, we successfully gained New York State Department of Health approval for a number of key products based on Enzo’s proprietary technology platforms. In November 2015, we announced approval of AMPIPROBE® HCV Assay for the quantitative detection of Hepatitis C. This assay is based on the proprietary nucleic acid amplification and detection technology platform which was the first in a line of products to be developed at Enzo to address the critical needs of the molecular diagnostics market and serves as validation of Enzo’s unique business strategy and structure.
In 2016, we were granted conditional approval of AMPIPROBE® Candidiasis Assay. This multiplex assay is designed to identify the presence of five of the most common species of Candida from a single vaginal swab. Industry estimates put the number of tests performed for the identification of Candida at over 10 million per year in the US alone. It is also estimated that over 70% of women will develop a Candida infection during their reproductive lifetime. While an independent assay, it will also serve as a component of a comprehensive women’s health panel.
In 2016, we were granted conditional approval of PLAQPRO™ Lp-PLA2 Assay. This is a biochemical activity assay designed to identify lipoprotein-associated phospholipase A2, a marker associated with the potential for coronary heart disease. The PLAQPRO™ Lp- PLA2 Assay can be useful as part of a cardiac testing panel for individuals at intermediate or high risk for developing coronary heart disease. Early identification of increased risk of developing coronary heart disease offers the opportunity to adjust patient lifestyles or utilize medical interventions to reduce risk. The assay was developed using the Company’s strong expertise in assay development, antibody production, small molecule chemistry, and detection technology. This cardiac assay delivers improved consistency and is designed to work on open platform clinical analysis instruments. The open platform configuration is one of the several factors that contribute to its cost effectiveness, which is vital to today’s clinical labs that are confronted by shrinking reimbursements.
In 2017, we were granted conditional approval for three additional women’s health related molecular diagnostic tests for use with the Company’s versatile and economic AMPIPROBE® platform. Approval was given for a real-time PCR-based method for qualitative detection of Neisseria gonorrhoeae, Chlamydia trachomatis and Trichomonas vaginalis in vaginal swab specimens. The Company’s AMPIPROBE®-based pipeline includes an extensive line of assays for identification of additional women’s health infectious diseases as well as for the quantification of viral load in serum or plasma specimens. This proprietary technology platform is the foundation of our ever-increasing line of medically relevant, cost-effective and easily adaptable solutions for clinical laboratories.
On October 23, 2017, we were granted conditional approval of another women’s health infectious disease diagnostic panel, which when combined with the Company’s previously approved panels, makes for one of the most comprehensive, efficient and affordable diagnostic products and services on the market today. A variety of infections, including sexually transmitted ones, are detected from a single vaginal swab collection via the Company’s proprietary, versatile and cost-effective AMPIPROBE® platform. The announcement results from recent action by the New York State Department of Health granting conditional approval of the new panel’s 5 additional tests, complementing the already approved 8 infectious disease tests for a total of 13 organisms being detectable from a single vaginal swab specimen.
On January 22, 2018, we were granted conditional approval of p16,
a marker used extensively as a key diagnostic and prognostic biomarker of several cancers. Enzo’s validated p16 provides
clear detection of tissue abnormalities in the field of cancer diagnostics, including cervical cancer’s progression. It complements
the company’s POLYVIEW® immunochemistry detection. With current mounting cost and reimbursement pressures, Enzo’s
new p16 test provides a highly cost-effective alternative. Other p16 tests on the market have of late become unaffordable as a
result of increasing reagent costs outweighing average reimbursements. When p16 is used in combination with Enzo’s POLYVIEW®
detection system’s reduction of false-positives, the economics are substantially enhanced. This and other similar compounds
comprise a $200 million market.
These assays are an important addition to Enzo’s expanding line of women’s health products, while also helping to solidify Enzo’s position as a leading full service women’s health lab.
Products in the Company’s development pipeline include an extensive line of assays for detection of numerous women’s health infectious agents as well as for use in the identification of pathogens for other markets. The Company also reported that it expects to roll-out a line of products designed to aid pathologists in distinguishing the characteristics of various tumors from biopsy specimens using technology developed by Enzo scientists. The Company’s molecular products are targeted at a market estimated to be in excess of $10 billion worth of laboratory service revenue.
Enzo is committed to delivering a robust line of products and services that will provide medically relevant, cost effective solutions that are easily adaptable to the workflow of clinical laboratories. The Company’s integrated Life Science and Clinical Lab structure continues to be instrumental in its ability to seamlessly develop and advance products from innovation and manufacturing in our life sciences group and validation and commercialization through our clinical laboratory. Our product development activity and pipeline include the following products:
| 7 |
| Expected Availability (1) | Platform | |
| HPV E6/E7 Detection | Available | FLOWSCRIPT® GENE EXPRESSION |
| HCV Viral Load | Available | AMPIPROBE® REAL-TIME AMPLIFICATION AND DETECTION |
| Cardiac Marker | Available | BIOCHEMICAL ASSAY |
| Fertility Assay | Q1 2019 | ENHANCED IMMUNOASSAY |
| Women’s Health Panel | Available | AMPIPROBE® REAL-TIME AMPLIFICATION AND DETECTION |
| HBV Viral Load | Q2 2019 | AMPIPROBE®REAL-TIME AMPLIFICATION AND DETECTION |
| HIV Viral Load | Q2 2019 | AMPIPROBE® REAL-TIME AMPLIFICATION AND DETECTION |
| IHC/ISH Detection | Available | ENHANCED DETECTION |
| FISH | Q1 2019 | DEEPSEE™ |
| Super CGH | Available | CYTAG® |
| TH1/TH2 | In development | ENHANCED IMMUNOASSAY |
| Cancer Marker Panel | Q3 2019 | In situ Hybridization ENHANCED DETECTION |
| HPV High Risk Panel | Q3 2019 | AMPIPROBE®REAL-TIME AMPLIFICATION AND DETECTION |
| HSV/VZV | Q3 2019 | AMPIPROBE®REAL-TIME |
| (1) | There can be no assurances these products can be successfully developed within these timeframes or available on these dates. |
Core Technologies
We have developed a portfolio of proprietary technologies with a variety of research, diagnostic and therapeutic applications.
Gene analysis technology
All gene-based testing is premised on the knowledge that DNA forms a double helix comprised of two complementary strands that match and bind to each other. If a complementary piece of DNA (a probe) is introduced into a sample containing its matching DNA, it will bind to, or hybridize, to form a double helix with that DNA. Gene-based testing is carried out by:
| • | amplification of the target DNA sequence (a process that is essential for the detection of very small amounts of nucleic acid); |
| • | labeling the probe with a marker that generates a detectable signal upon hybridization; |
| • | addition of the probe to the sample containing the DNA; and |
| • | binding or hybridization of the probe to the target DNA sequence, if present, to generate a detectable signal. |
We have developed the AMPIPROBE® platform as a broad technology base for the labeling, detection, amplification and formatting of nucleic acids for gene analysis which is supported by our significant proprietary position in these fields. This and other proprietary technologies are the building blocks of our Molecular Diagnostics platforms.
| 8 |
Amplification
In the early stages of infection, a pathogen may be present in very small amounts and consequently may be difficult to detect. Using DNA amplification, samples can be treated to cause a pathogen’s DNA to be replicated, or amplified, to detectable levels. We have developed a proprietary amplification process for multicopy production of nucleic acids, as well as proprietary techniques for amplifying the signals of our probes to further improve sensitivity. Our amplification technologies are particularly useful for the early detection of very small amounts of target DNA. We have also developed isothermal amplification procedures that can be performed at constant temperatures, unlike polymerase chain reaction (PCR) the most commonly used method of target nucleic acid amplification. These platform technologies could thus potentially lead to assays with advantages over PCR-based tests which require expensive heating and cooling systems or specialized heat-resistant enzymes. Moreover, our AMPIPROBE® Nucleic Acid Amplification Platform, because of the reduced amount of starting material needed for analysis, may lead to a next-generation of molecular diagnostics that can impart higher sensitivity at a lower cost than currently available assays.
Flow Cytometry
We have developed and launched our first product using our proprietary FLOWSCRIPT® platform using flow cytometry to analyze messenger RNA (mRNA) transcript expression in individual cells in a mixed cell population. By studying whether a gene or a set of genes is turned on or off, it is possible to obtain clinically relevant information at the single cell level. Our first product, the FLOWSCRIPT® HPV E6/E7 Assay, examines the levels of E6/E7 mRNA transcripts from multiple high risk types which account for over 95% of cervical cancers. We are planning to develop and introduce other products based on this platform technology in the future for applications such as immune-mediated disorders, metabolic disorder patient monitoring, and other cancers.
Non-radioactive labelling and detection
Traditionally, nucleic acid probes were labeled with radioactive isotopes. However, radioactively labeled probes have a number of shortcomings; they are unstable and consequently have a limited shelf life and they are potentially hazardous, resulting in restrictive licensing requirements and safety precautions for preparation, use and disposal. Finally, radioactive components are expensive. Our technologies permit gene analysis without the problems associated with radioactively labeled probes and are adaptable to a wide variety of formats.
Formats
There are various processes, or formats, for performing probe-based tests. In certain formats, the probe is introduced to a target sample affixed to a solid matrix; in others, the probe is combined with the sample in solution (homogeneous assay). Solid matrix assays include: in situ assays in which the probe reaction takes place directly on a microscope slide; dot blot assays in which the target DNA is fixed to a membrane; and microplate and microarray assays in which the DNA is fixed on a solid surface, and the reaction can be quantified by instrumentation.
Therapeutic Platform Development
Cell Signaling Pathway
One area of Enzo’s therapeutic platform development is related to the development of pharmaceutical agents that affect protein-protein interactions. Over the past several years, our scientists and collaborators have unlocked the secrets of a major cell signaling pathway, thus producing a means to modify biological activity in a number of physiological systems.
Further investigation into the design and control of this system has allowed our scientists and their collaborators to determine the structure of key regulatory proteins and to identify active sites that can then become targets for Enzo’s proprietary technology generating system. Our technology is capable of generating active compounds that range from orally delivered small molecules to peptides, oligonucleotides and antibodies. We have performed pioneering work on the structure and function of lipoprotein receptor-related protein (LRP) and its ligands, developed a screening technology to identify active compounds, and synthesized proprietary molecules capable of producing biological effects in cell-based systems and animal models of disease. Specifically, this system allows the Company to:
| • | generate biological, genetic, and structural information concerning LRP; |
| • | determine the structure of LRP docking sites of its ligands; |
| • | identify the functionally important residues via site-directed mutagenesis; |
| 9 |
| • | build the fine structure map and employ it as the basis for virtual screening; |
| • | show that compounds specifically bind to wild type LRP5, but not to mutated LRP5; |
| • | generate a cell-based assay capable of identifying active compounds; and |
| • | synthesize proprietary molecules that are active in animal models of disease. |
Through this novel, proprietary, functional screening system, we have identified small molecules capable of reversing sclerostin-mediated inhibition of Wnt signalling. Preclinical animal studies with several candidate lead compounds produced the following results:
| • | significant increases in total and femoral bone density through new bone formation; |
| • | significant reduction in alveolar bone loss; and |
| • | significant reduction in bone resorption. |
The anabolic induction of new bone formation and prevention of bone loss by our small molecule compounds may promise new paths for the treatment of osteoporosis. In addition, our proprietary technology has enabled the generation of novel chemical entities that have significant glucose lowering activity. These effects are separate from its effects on bone metabolism indicating a specificity of action conferred by the interaction of a particular compound with the cell signaling pathway. Therefore, this approach may be broadly applicable to the generation of therapeutic drug candidates for multiple indications.
Oral Immune Regulation
We continue to explore a novel therapeutic approach based on immune regulation. Our immune regulation technology seeks to control an individual’s immune response to a specific antigen in the body. An antigen is a substance that the body perceives as foreign and, consequently, against which the body mounts an immune response. This platform technology is being developed as a means to manage immune-mediated diseases, such as autoimmune uveitis and Crohn’s disease.
We have developed an immunomodulation agent EGS21 as a potential therapeutic for treating immune-mediated disorders. EGS 21 is a glycolipid that has been shown by our scientists and collaborators to act as an anti-inflammatory agent in animal model systems and is being evaluated as a drug candidate in the treatment of various immune-mediated diseases.
Gene Regulation
We have developed an approach to gene regulation known as genetic antisense or antisense RNA. Our technology involves the introduction of a gene into cellular DNA that codes for an RNA molecule that binds to, and thus deactivates, RNA produced by a specific gene. To deliver our antisense gene to the target cell in a process called transduction, we have developed proprietary vector technology.
We believe, though there can be no assurance, that our vector technology has broad applicability in the field of gene medicine. This can be attributed to the following properties of our construct:
| • | the viral promoters are inactivated; |
| • | insertional gene activation is prevented - a major safety factor; |
| • | chromosomal integration; and |
| • | nuclear localization. |
There can be no assurance that we will be able to secure patents or that these programs will be successful. The potential therapies we are developing could be used, if successful for the treatment of a variety of diseases, including osteoporosis, osteonecrosis and other bone pathologies, diabetes, autoimmune uveitis and inflammatory bowel disease, including Crohn’s disease and ulcerative colitis, among others.
| 10 |
Clinical Laboratory Services
We operate a regional clinical laboratory that offers extensive diagnostic services to the New York and New Jersey medical communities. As part of our ongoing strategic growth plan we have recently expanded service to Connecticut and New England states. Our clinical laboratory testing is utilized by physicians as an essential element in the delivery of healthcare services. Physicians use laboratory tests to assist in the detection, diagnosis, evaluation, monitoring and treatment of diseases and other medical conditions. Clinical laboratory testing is generally categorized as clinical testing or anatomic pathology testing. Clinical testing is performed on body fluids, such as blood and urine. Anatomical pathology testing is performed on tissues and other samples, such as human cells. Many clinical laboratory tests are considered routine and can be performed by most commercial clinical laboratories.
Tests that are not routine and that require more sophisticated equipment and highly skilled personnel are considered esoteric tests and may be performed less frequently than routine tests.
We offer a comprehensive and broad range of routine esoteric and molecular diagnostic clinical laboratory tests or procedures. These tests are frequently used in general patient care by physicians to establish or support a diagnosis, to monitor treatment or medication levels, or to search for an otherwise undiagnosed condition.
Our full service clinical laboratory in Farmingdale, New York contains an infrastructure that includes comprehensive information technology applications, logistics, client services and billing departments. We have a network of over thirty strategically located patient service centers and a full service phlebotomy department. Patient service centers collect from patients the specimens as requested by physicians. We also operate a fully equipped STAT laboratory in New York City. A “STAT” lab has the ability to perform certain routine tests quickly and report results to the physician immediately.
Patient specimens are delivered to our laboratory facilities primarily by our logistics department accompanied by a test requisition form. These forms, which are completed by the ordering physician, indicate the tests to be performed and demographic patient information and in most instances are transmitted to us via EnzoDirect, our proprietary computer-based ordering and results delivery system. Once the information is entered into the laboratory computer system the tests are performed on the corresponding laboratory testing instrumentation and the results are uploaded primarily through an interface from the laboratory testing instrumentation or in some instances, manually entered into the laboratory computer system. Most routine testing is completed by early the next morning, and test results are reported to the ordering physician. These test results are either reported electronically via EnzoDirect to a physician office Electronic Medical Records (EMR) system or delivered by our logistics department directly to the ordering physicians’ offices. Physicians who request that they be called with a particular result are accordingly notified by our customer service personnel.
For fiscal years ended July 31, 2018, 2017 and 2016, respectively, approximately 71%, 72% and 69% of the Company’s revenues were derived from the clinical laboratory services. Revenues, net of contractual adjustment, from direct billings under the Federal Medicare program during the years ended July 31, 2018, 2017 and 2016 were approximately 16% of the clinical laboratory services segment’s total revenue. The contractual adjustment is an estimate that reduces gross revenue, based on gross billing rates, to amounts expected to be approved and reimbursed. We estimate contractual adjustment based on significant assumptions and judgments, such as the interpretation of payer reimbursement policies which bears the risk of change. The estimation process is based on the experience of amounts approved as reimbursable and ultimately settled by payers, versus the corresponding gross amount billed to the respective payers. Other than the Medicare program, revenues from UnitedHealthcare and Oxford Health Plan represented approximately 39%, 39% and 30% of the Clinical Laboratory Services segment’s net revenue for the fiscal year ended July 31, 2018, 2017 and 2016, respectively.
At July 31, 2018 and 2017, approximately 74% and 75% respectively of the Company’s net accounts receivable was derived from its clinical laboratory business. The Company believes that the concentration of credit risk with respect to the Clinical Labs accounts receivable is mitigated by the diversity of its third party payers that insure individuals. To reduce risk, the Company routinely assesses the financial strength of these payers and, consequently, believes that its accounts receivable credit risk exposure, with respect to these payers, is limited. While the Company also has receivables due from the Federal Medicare program, the Company does not believe that these receivables represent a credit risk since the Medicare program is funded by the federal government and payment is primarily dependent on our submitting the appropriate documentation.
Gross billings are based on a standard fee schedule we set for self-payers, all third party payers, including Medicare, health maintenance organizations (“HMO’s) and managed care providers and expanding institutional relationships with direct billing. We adjust the contractual adjustment estimate quarterly, based on our evaluation of current and historical settlement experience with payers, industry reimbursement trends, and other relevant factors. The other relevant factors that affect our contractual adjustment include the monthly and quarterly review of: 1) current gross billings and receivables and reimbursement by payer, 2) current changes in third party arrangements, and 3) the growth of in-network provider arrangements and managed care plans specific to our Company. The clinical laboratory industry is characterized by a significant amount of uncollectible accounts receivable related to the inability to
| 11 |
receive accurate and timely billing information in order to forward it on to the third party payers for reimbursement, and the inaccurate information received from the covered individual patients for unreimbursed unpaid amounts.
Billing for laboratory services is complicated. Depending on the billing arrangement and applicable law, we must bill various payers, such as patients, insurance companies and the Federal Medicare Program, all of which have different requirements. In both New York and New Jersey, the law prohibits the Company from billing the ordering physician. Compliance with applicable laws and regulations, as well as internal compliance policies and procedures add further complexity to the billing process. We depend on the ordering physician to provide timely, accurate billing demographic and diagnostic coding information to us. Additional factors complicating the billing process include:
| • | pricing differences between our standard gross fee schedules and the reimbursement rates of the payers; |
| • | disputes with payers as to which party is responsible for payment; |
| • |
disparity in coverage and information requirements among various payers; and
|
| • | differences in medical policies established by various payers. |
Most of our bad debt expense is primarily the result of inaccurate billing information on requisitions received from the ordering physician. In addition, the bad debts includes the balances, after receipt of the approved settlements from third party payers for the insufficient diagnosis information received from the ordering physician, which result in denials of payment and the uncollectible portion of receivables from self-payers, including deductibles and co-payments, which are subject to credit risk and patients’ ability to pay. We must perform the requested tests and report test results regardless of whether the billing or diagnostic coding information is inaccurate or missing. We subsequently attempt to contact the ordering physician to obtain and rectify incorrect billing information. Missing or inaccurate information on the requisitions adds complexity to and may slow the billing process, creates backlogs of unbilled requisitions, and generally decreases the collectability and increases the aging of accounts receivable. When all issues relating to the missing or inaccurate information are not resolved in a timely manner, the related receivables are fully reserved to the allowance for doubtful accounts or allowances for contractual adjustments or written off.
We incur significant additional costs as a result of our participation in Medicare, as billing and reimbursement for clinical laboratory testing is subject to considerable and complex and stringent federal and state regulations including those relating to coverage, billing and reimbursements. Future changes in regulations could further complicate our billing and increase our billing expenses. These additional costs include those related to: (1) complexity added to our billing processes and changes to our reimbursements; (2) training and education of our employees and customers; (3) compliance and legal costs; and (4) costs related to, among other factors, medical necessity denials and advance beneficiary notices. The Centers for Medicare & Medicaid Services, or CMS (formerly the Health Care Financing Administration), establishes procedures and continuously evaluates and implements changes in the reimbursement process.
The established Medicare reimbursement rate for clinical laboratory services has been reduced by the Federal government in a number of instances over the past several years. In March 2010, U.S. federal legislation was enacted to reform healthcare. The legislation provides for reductions in the Medicare clinical laboratory fee schedule of 1.9% for five years beginning in 2010 and also includes a productivity adjustment which reduces the Consumer Price Index (“CPI”) market basket update beginning in 2011. Based on these calculations, the Medicare Fee Schedule was unchanged in calendar years 2016 and 2017, and decreased in 2018.
The Patient Protection and Affordable Care Act imposes an excise tax on the seller for the sale of certain medical devices in the United States, including those purchased and used by laboratories, beginning in 2013 and establishes the Independent Payment Advisory Board (“IPAB”). If the projected growth in per capita Medicare costs exceeds a specified target level, the IPAB must submit proposals to reduce or eliminate the difference. For calendar years 2016 through 2019, the target growth rate is the projected average of the increases in the Consumer Price Index and the medical care expenditure category of the Consumer Price Index; for 2020 and thereafter, the target growth rate is the rate of increase in gross domestic product per capita plus one percentage point. If it is necessary for the IPAB to submit proposals, they will automatically be implemented unless Congress enacts alternative proposals that achieve the same savings targets. We could experience a significant decrease in revenue from Medicare as a result of these pieces of legislation, which could have a material adverse effect on us. The IPAB currently has no appointees and it is unclear whether when and if it will become operational.
Diagnostic Products
We are a manufacturer of labeling and detection technologies from DNA to whole cell analysis. Enzo’s products are backed by innovative technology platforms and a deep patent portfolio. With 40 years of experience, Enzo continues to provide integrated solutions for drug development, pipeline basic research, drug discovery, quality control in drug development and diagnostics. Enzo Life Sciences offers a broad range of high-quality products to advance research including proteins, antibodies, peptides, small
| 12 |
molecules, labeling probes, dyes, and kits. Enzo operates in a highly competitive and price-sensitive marketplace and is repositioning itself by narrowing its product mix to concentrate on improved profitability, while also adding staff who are more experienced in operations. We have become a specialized assay supplier as part of our integrated strategic plan to deliver highly efficient, cost-effective assays for our own use and to sell to independent labs. With direct sales operations in the US, Switzerland, Germany, UK, France, and Benelux, Enzo Life Sciences also supports its products through a global network of dedicated distributors.
With a passion for genomics, Enzo was the first to develop products for non-radioactive labeling of nucleic acids. This technique was instrumental in the development of today’s genomic analysis market. Our pioneering research in genetic modification medicine was the first to recognize that nucleic acids could be used as therapeutics. Our innovations in the detection of nucleic acids in solutions and solid matrices led to the development of technology platforms such as hybrid capture, as well as fluorescent and chromogenic in situ hybridization. Enzo remains at the forefront of target amplification technologies critical in the detection of infectious agents, cancer markers, and genotyping. Our work in the genomic space has resulted in technologies in gene expression and immune system regulation, which opened the door for the well-known molecular diagnostics assays used today.
The products produced and supplied include small molecules, proteins, antibodies, peptides, probes, assay kits and custom services. Our comprehensive portfolio of high quality reagents and kits in key research areas are sold to scientific experts in the following fields:
| Adipokines | Interferons | |
| Antibiotics | In situ Hybridization | |
| Autophagy/Apoptosis/Cell Death | Kinases/Inhibitors | |
| Biologically Active Peptides | Leukotrienes/Prostaglandins/Thromboxanes | |
| Bone Metabolism | Microarray Labeling | |
| Cancer Research | Multidrug Resistance | |
| Cell Death | Natural Products/Antibiotics | |
| Cell Cycle | Neuroscience Research | |
| Chemokines/Cytokines | Nitric Oxide Pathway | |
| Cytoskeletal Research | Nuclear Receptors | |
| Dependence Receptors | Oxidative Stress | |
| DNA Fragmentation/Damage/Repair | Protein Aggregation | |
| DNA Regulation | Proteosome/Ubiquitin | |
| Epigenetics | Receptors | |
| FISH | Signal Transduction | |
| Growth Factors/Cytokines | Stem Cell/Cell Differentiation | |
| Hypoxia | Stress Proteins/Heat Shock Proteins | |
| Immunology | Toxicology | |
| Immunohistochemistry | TNF/TNF Receptor Superfamily | |
| Viral Signaling | Transcription Factors | |
| Inflammation/Innate Immunity | WNT Research |
We maintain acquired brands including Alexis, Biomol International, Assay Designs, and Stressgen. Enzo strategically uses these brands to complete our product portfolio, allowing us to offer complete solutions to researchers in all fields. These brands are complementary to our core expertise in genomics and molecular biology. The Company intends to maintain the rights to the acquired brands which have long product histories. The Company believes the emphasis on the Enzo Life Sciences brand will result in stronger and clearer brand awareness and allow the Company to execute the sale of higher value products and promote more products into the drug development, clinical research and diagnostic markets.
Axxora.com -“The Reagents Marketplace”, Thousands of Reagents, One Marketplace Axxora.com is a proven distribution platform for original manufacturers of innovative research reagents. An increasing number of researchers use our unique marketplace to connect with over 40 specialty manufacturers and gain access to over 40,000 products.
Research and Development
Our principal research and development efforts are directed toward developing innovative new clinical research and diagnostic platforms, and selective expansion of our research product lines, given our manufacturing and distribution capability. We have developed our core research expertise in the life sciences field as a result of over 40 years of dedicated focus in this area. We conduct our research and other product development efforts through internal research and collaborative relationships.
In the fiscal years ended July 31, 2018, 2017 and 2016, the Company incurred costs of approximately $3.2 million, $2.9 million, and $3.5 million, respectively, for research and development activities. During fiscal 2018, the Company’s research and development
| 13 |
program was refocused to areas that had greater opportunity in molecular diagnostics and immunology chemistry to maximize revenues.
Internal Research Programs
Our professional staff, including 39 with post graduate degrees, performs our internal research and development activities. Our product development programs incorporate various scientific areas of expertise, including recombinant DNA, monoclonal antibody development, enzymology, microbiology, biochemistry, molecular biology, organic chemistry, immunology, flow cytometry and fermentation. In addition, we continuously review in-licensing opportunities in connection with new technology.
External Research Collaborations
We have and continue to explore collaborative relationships with prominent companies and leading-edge research institutions in order to maximize the application of our technology in areas where we believe such relationship will benefit the development of our technology.
Sales and Marketing
Our sales and marketing strategy is to sell our life sciences products through: (i) direct sales to end-users under the Enzo Life Sciences name, with direct recognition to our acquired brands (ii) direct sales to end users under the Axxora electronic market place name (iii) supply agreements with manufacturers and (iv) distributors in major geographic markets. We operate with an understanding of local markets and a well-functioning distribution network system across the globe. Scientists around the world who recognize the brands (Alexis, Assay Designs, Biomol, Enzo and Stressgen) now receive products directly from Enzo Life Sciences where we are recognized for innovative high quality products, supported directly by our qualified technical staff. We sell the same products through our Axxora electronic market place which is also the source for life science research reagents from over 40 original manufacturers. Our direct marketing and sales network includes fully-owned subsidiaries (USA, Switzerland, Germany, Benelux, and UK), a branch office in France and a network of third party distributors in most other significant markets worldwide.
For Diagnostic Services, we focus our sales efforts on obtaining and retaining profitable accounts. We market the clinical laboratory services to a broad range of ordering physicians in the metro New York, New Jersey and Connecticut regions through our direct sales force who are supported by client service and patient service representatives. We monitor and where appropriate, change the service levels and terminate ordering physician accounts that are not profitable. We are focusing our efforts to attract and retain clients who participate with the providers with whom we have regional contracts and are consistently looking to add higher value molecular and esoteric testing, both internally developed and with partners, to our menu to assist sales in new account penetration as well as to improve our level of service to existing clients.
Distribution Arrangements
We also distribute our life science products internationally through a network of distributors. Through these arrangements, we are able to leverage the established marketing and distribution infrastructure of these companies in certain market places.
Competition
We compete with other life science and biotechnology companies, as well as pharmaceutical, chemical and other companies. Competition in our industry is intense. Many of these companies are performing research targeting the same technologies, applications and markets. Many of these competitors are significantly larger than we are and have more resources. The primary competitive factors in our industry are the ability to create scientifically advanced technology, offer innovative products at the forefront of technological development to targeted market segments, successfully develop and commercialize products on a timely basis, establish and maintain intellectual property rights and attract and retain a breadth and depth of human resources.
Our clinical laboratory services business competes with numerous national, regional, and local entities, some of which are larger than we are and have greater financial resources than we do. Our laboratory competes primarily on the basis of the quality and specialized nature of its testing, reporting and information services, its reputation in the medical community, its reliability and speed in performing diagnostic tests, and its ability to employ qualified laboratory personnel.
Intellectual Property
We consider our intellectual property program to be a key asset and a major strategic component to the execution of our business strategy. A broad portfolio of issued patents and pending patent applications supports our core technology platforms. Our policy is to
| 14 |
seek patent protection for our core technology platforms, as well as for ancillary technologies that support these platforms and provide a competitive advantage.
At the end of fiscal 2018 we owned or licensed 343 patents relating to products, methods and procedures resulting from our internal or sponsored research projects. There can be no assurance that patents will be issued on pending applications or that any issued patents will not be challenged (see Item 3, Legal Proceedings), or that they will have commercial benefit. We do not intend to rely on patent protection as the sole basis for protecting our proprietary technology. We also rely on our trade secrets and continuing technological innovation. We require each of our employees to sign a confidentiality agreement that prohibits the employee from disclosing any confidential information about us, including our technology or trade secrets.
Our intellectual property portfolio can be divided into patents that provide claims in three primary categories, as described below:
Nucleic Acid Chemistry
We currently have broad patent coverage in the area of nucleic acid chemistry. We have done extensive work on the labeling of nucleic acids for the purpose of generating a signal that dates back over twenty years. Enzo has multiple issued patents covering the modification of nucleic acids at their sugar and phosphate sites. The claims contained in these patents cover products that incorporate a signaling moiety into a nucleic acid attached to a sugar or phosphate for the purpose of nucleic acid detection or quantification, including sequencing and real time nucleic acid amplification. Enzo also has patents directed to proprietary dyes that may be used to label the sugar, base or phosphate positions of nucleic acids.
Signal Delivery
We also have a long history of innovation in the area of analyte detection using non-radioactive signaling entities. At the signaling entity itself, there are several Enzo patents that cover the formation of this structure. A patent which was allowed in 2006 covers the attachment of signaling molecules through the phosphate moiety of a nucleic acid, which is how the signal-generating enzyme is bound.
Nucleic Acid Analysis Format
We also have patents with issued claims covering the use of arrays of single-stranded nucleic acids fixed or immobilized in hybridizable form to a non-porous solid support. These patents cover any product that uses arrays of nucleic acids for molecular analysis.
In some instances, we may enter into royalty agreements with collaborating research parties in consideration for the commercial use by us of the developments of their joint research. In other instances the collaborating party might obtain a patent, but we receive the license to use the patented subject matter. In such cases, we will seek to secure exclusive licenses. In other instances, we might have an obligation to pay royalties to or reach a royalty arrangement with a third party in consideration of our use of developments of such third party.
REGULATION AFFECTING OUR BUSINESSES
Clinical Laboratory Services
The clinical laboratory industry is subject to significant federal and state regulation, including inspections and audits by governmental agencies. Governmental authorities may impose fines, criminal penalties or take other actions to enforce laws and regulations, including, but not limited to, revocation of a clinical laboratory’s certificate and/or license to operate a clinical laboratory. Changes in regulation may also increase the cost of performing clinical laboratory tests, increase administrative requirements, or decrease the amount of reimbursement. Our clinical laboratory and (where applicable) patient service centers are licensed and accredited as required by law.
CLIA (The Clinical Laboratory Improvement Act of 1967, and the Clinical Laboratory Improvement Amendments of 1988) regulates virtually all clinical laboratories in the United States. Among other things, CLIA requires laboratories to earn certification from the federal government and comply with various operational, personnel and quality requirements intended to ensure that their clinical laboratory testing services are accurate, reliable and timely. CLIA does not preempt state laws that are more stringent than federal laws. As such, certain clinical laboratories must meet state specific standards and undergo proficiency testing and inspections. Clinical laboratory certificates or licenses are also required by various state and local laws.
CLIA assigns test into one of three categories on the basis of complexity (waived, moderate complexity and high complexity) and establishes varying requirements depending upon the complexity category of the test performed. A laboratory that performs high
| 15 |
complexity tests must meet more stringent requirements than a laboratory that performs only moderate complexity tests, while those that perform only waived tests may apply for a certificate of waiver that if granted, would exempt the laboratory from most CLIA requirements. Our facility is certified to perform high complexity tests. In general, regulations promulgated by the United States Department of Health and Human Services (“HHS”) require laboratories that perform high or moderate complexity tests to implement systems that ensure the accurate performance and reporting of test results, establish quality control and quality assurance systems, ensure that personnel meet specified standards, conduct proficiency testing by approved agencies, and undergo biennial inspections, among other requirements.
Clinical laboratories also are subject to state regulation. CLIA provides that a state may adopt different or more stringent regulations than Federal law, and permits states to apply for exemption from CLIA if HHS determines that the state’s laboratory laws are equivalent to, or more stringent than, CLIA. The State of New York’s clinical laboratory regulations contain provisions that are more stringent than Federal law, and New York has received exemption from CLIA. Therefore, as long as New York maintains a licensure program that is CLIA-exempt, laboratories in New York, including our laboratory, are regulated under New York law rather than CLIA. Our laboratory is licensed in New York and has continuing programs to ensure that its operations are in compliance with all applicable regulatory requirements.
Sanctions for non-compliance with applicable regulations may include, but are not limited to, suspension, revocation, or limitation of a laboratory’s CLIA certificate or state license, as well as fines and criminal penalties. The loss of, or adverse action against, a certificate or license, the imposition of fines, penalties or other sanctions, or future changes in Federal, state or local laboratory laws and regulations (or in the interpretation of current laws and regulations) could have a material adverse effect on our business.
Billing and reimbursement for clinical laboratory testing is subject to complex federal and state laws, rules and regulations, the violation of which may include, but is not necessarily limited to: (1) exclusion from participation in federal health care programs (including Medicare and Medicaid); (2) asset forfeitures; (3) civil monetary penalties; (4) criminal fines and penalties; and (5) the loss of licenses, certificates and/or authorizations necessary to operate some or all of a clinical laboratory’s business.
The health care industry has been undergoing significant change because third-party payers, such as Medicare, Medicaid, health maintenance organizations and commercial insurers, have increased their efforts to control the cost, utilization and delivery of health care services. To address the problem of increasing health care costs, legislation has been proposed or enacted at both the Federal and state levels to regulate health care delivery in general, and clinical laboratories in particular. Additional health care reform efforts are likely to be proposed in the future. In particular, we believe that reductions in reimbursement for Medicare services will continue to be implemented from time to time. Reductions in the reimbursement rates of other third-party payers, commercial insurer and health maintenance organizations are likely to occur as well. We cannot predict the effect that current and future health care reform measures, if enacted, would have on our business, and there can be no assurance that such reforms, if enacted, would not have a material adverse effect on our business and operations.
Containment of health care costs, including reimbursement for clinical laboratory services, has been a focus of on-going governmental activity. Clinical laboratories must bill Medicare directly for the services provided to Medicare beneficiaries and may only collect the amounts permitted under the Medicare Clinical Laboratory Fee Schedule. Under the Patient Protection and Affordable Care Act, expansion in the pool of covered lives may expand the market for clinical diagnosis testing while at the same time, various policies aimed at reducing costs or bundling care may reduce the rates paid for such services; the net impact of these factors on the market for our tests is not clear . In April 2014, Congress passed the Protecting Access to Medicare Act of 2014 (PAMA), which included substantial changes to the way in which clinical laboratory services will be paid under Medicare. Beginning in 2018, Medicare payments for clinical laboratory services are paid based upon private payer rates as reported by clinical laboratories across the US replacing the current system which is based upon fee schedules derived from historical charges for tests from approximately 30 years ago. The final regulation to implement Medicare laboratory payment reform was released on June 17, 2016 by CMS. Since Enzo’s clinical lab receives more than 50% of its total Medicare revenue from the Part B Clinical Laboratory Fee Schedule and the Physician Fee Schedule and receives more than $12,500 in Medicare revenues per year, we are considered an “applicable laboratory”, and as such, reported private payer fee reimbursements for the period January 1, 2016 to June 30, 2016 to CMS by March 31, 2017. This data was aggregated and utilized as the basis for the 2018 fee schedules that were finalized in November 2017.
Future changes in federal, state and local regulations (or in the interpretation of current regulations) affecting governmental reimbursement for clinical laboratory testing could have a material adverse effect on our business. We cannot predict, however, whether and what type of legislation will be enacted into law. In addition, reimbursement disapprovals by the third party payers, commercial insurers and health maintenance organizations, reductions or delays in the establishment of reimbursement rates, carrier limitations on the insurance coverage of the Company’s services or the use of the Company as a service provider could have a negative effect on the Company’s future revenues. During our fiscal 2016 and 2017, reimbursement rates remained constant with 2015 levels. However, PAMA reimbursed cuts impacted fiscal 2018 results beginning in January 2018. PAMA cuts are mandated for 2019 and 2020 and are expected to impact our fiscal 2019 and 2020 results.
| 16 |
Anti Fraud and Abuse Laws
Existing Federal and state laws also regulate certain aspects of the relationship among healthcare providers, including clinical laboratories, and their referral sources (i.e., physicians, hospitals, other laboratories, etc.). One of these laws, known as the “Anti-Kickback Statute,” contains extremely broad prohibitions against giving, accepting, soliciting (i.e., asking for) or arranging for remuneration in any form (i.e., cash, gifts, certain discounts, cross-referrals between parties, etc.), either directly or indirectly, for the purpose of inducing or rewarding another party for referrals of items or services paid for by a federal government health care program. The Anti-Kickback statute is very broad and includes the purchasing, ordering, leasing or arranging for, or recommending the purchase, leasing or ordering of, services paid for by a federal health care program in exchange for remuneration (i.e., anything of value).
Violation of the Anti-Kickback Statute may result in, among other things, a criminal conviction, significant monetary penalties and exclusion from federal health care programs (including Medicare and Medicaid). Any person or entity involved in a prohibited transaction is potentially subject to criminal and civil penalties. A laboratory that claims payment for business generated by the Anti-Kickback Statute may also be subject to prosecution for violating a separate civil statute, the federal False Claims Act.
The False Claims Act is also a broad statute that the government often utilizes to combat fraud and abuse in the health care environment. Among other things, the statute is violated by any person who knowingly presents, or causes to be presented, a false or fraudulent claim for payment or approval; knowingly makes, uses, or causes to be made or used, a false record or statement material to a false or fraudulent claim; conspires to commit the above (or other specified) violations; or knowingly makes, uses, or causes to be made or used, a false record or statement material to an obligation to pay or transmit money or property to the government, or knowingly conceals or knowingly and improperly avoids or decreases an obligation to pay or transmit money or property to the government. The False Claims Act also provides that private parties may bring an action on behalf of (and in the name of) the United States to prosecute a False Claims Act violation. These private parties (known as “qui tam relators”) may share in a percentage of the proceeds that result from a False Claims Act action or settlement. A person or entity found to have violated the False Claims Act may be held liable for a per claim civil penalty of not less than $5,500 and not more than $11,000, plus three times the amount of damages sustained by the government. A person violating the False Claims Act is also liable for the costs of the civil action brought to recover any such penalty or damages. Other consequences may also result from a violation of the False Claims Act. New York has also adopted its own False Claims Act statute, which closely mirrors its federal counterpart.
Another Federal law, commonly known as the “Stark” law, prohibits physicians who have a financial relationship with an entity that furnishes “designated health services,” which includes clinical laboratory services (including anatomic pathology and clinical chemistry services), from referring Medicare (and in certain instances Medicaid) beneficiaries to that entity for laboratory tests unless a specific exception applies.
In addition, laboratories may not bill federal health care programs, or any other payer, for services furnished pursuant to a prohibited referral. Violation of the Stark law may result not only in denial of payment for the underlying testing services, but also the imposition of civil monetary penalties and, potentially, False Claims Act liability. New York State has adopted laws that are similar to the Federal Stark law, which contain similar prohibitions and penalties and apply regardless of payer.
The Stark law and New York State regulations have also placed restrictions on the supplies and other items that laboratories may provide to their clients. These laws specify that laboratories may only provide clients with items or devices that are used solely to collect, transport or store specimens for the laboratory or to communicate results or tests. Items such as biopsy needles, snares and reusable needles are specifically prohibited from being supplied by laboratories to their clients. The Company has implemented procedures to ensure compliance with these laws and restrictions.
In February 1997, the Department of Health and Human Services, Office of the Inspector General (OIG) released model voluntary compliance program guidance for laboratories. One key aspect of the model compliance guidance was an emphasis on the responsibility of laboratories to notify physicians that Medicare covers only medically necessary services. This requirement, and the likely effect on physician test ordering habits, focuses on chemistry tests, especially routine tests, rather than on anatomic pathology services or the non-automated tests, which make up the majority of the Company’s business measured in terms of net revenues. Nevertheless, it could potentially affect physicians’ test ordering habits more broadly. The Company is unable to predict whether, or to what extent, these developments have impacted, or may impact, utilization of the Company’s services.
The federal health care reform legislation adopted in March, 2010, known as the Patient Protection and Affordable Care Act, contains provisions requiring providers to establish compliance programs as a condition of enrollment in Medicare, Medicaid and the State Children’s Health Insurance Program. Implementing regulations and guidance for clinical laboratories has not yet been issued yet by the Centers for Medicare and Medicaid Services. In addition, New York State has adopted mandatory compliance program requirements for certain specified providers, including those who directly or indirectly bill or collect more than $500,000 annually in Medicaid payments, and entities licensed under certain articles of the Public Health Law and Mental Hygiene Law, respectively. The Company has adopted its own Corporate Compliance Program based upon the OIG model program guidance and in accordance with New York State’s requirements.
| 17 |
The Company’s compliance program focuses on, among other things, establishing clear compliance standards; auditing and monitoring of the Company’s billing and coding practices; training personnel on compliance standards, policies and procedures; preventing and detecting fraud, waste and abuse, enforcing a policy of non-retaliation and non-intimidation for good faith participation in the compliance program; and establishing good faith reporting of actual or suspected compliance violations.
The Company seeks to structure its arrangements with physicians and other customers in compliance with federal and state Anti-Kickback laws, Stark laws, False Claims Acts, and other applicable laws, rules and regulations, and to keep current on developments concerning their application to the Company, including consultation with legal counsel. However, the Company is unable to predict how such laws and regulations will be interpreted and applied in the future, and thus no assurances can be given that its arrangements or processes will not become subject to scrutiny by a governmental agency.
Confidentiality of Health Information
The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) included “administrative simplification” provisions designed to standardize common electronic transactions in health care and to protect the security and privacy of health information. Congress’ purpose in promulgating HIPAA was to increase the efficiency of health care transactions while, at the same time, protecting the confidentiality of patient information. Regulations have been adopted for electronic transaction, privacy security and breach notification standards and include the requirement to use a National Provider Identifier in electronic health care transactions. The National Provider Identifier is an identifier that replaced all other identifiers that are currently used or healthcare transactions (e.g., UPIN, Medicaid provider numbers, identifiers assigned by commercial insurers). The regulations promulgated under HIPAA have very broad applicability, including by specifically applying to health care providers, which include physicians and clinical laboratories that conduct an electronic transaction for which HIPAA has articulated standards. Together, health plans, health care clearinghouses and health care providers that conduct standard transactions subject to HIPAA are referred to as “Covered Entities”.
The electronic transaction standards regulations created guidelines for certain common health care transactions. With certain exceptions, these standards require that, when we conduct certain transactions electronically with another health care provider, health care clearinghouse or health plan, we must comply with the standards set forth in the regulations. The regulations established standard data content and format for submitting electronic claims and other administrative health transactions. Health care providers and health plans are required to use standard formats when transmitting claims, referrals, authorizations, and certain other transactions electronically. The Company believes it is in compliance with these standards.
Privacy, security and breach notification requirements regarding protected health information (“PHI”).
We are required to maintain numerous policies and procedures in order to comply with the HIPAA privacy security and breach notification requirements. Furthermore, we need to continuously ensure that there are mechanisms in place to safeguard the privacy of PHI that is transmitted or maintained in any format (e.g. oral, written, or electronic). Failure to comply with these requirements can result in criminal and civil penalties. To comply with the HIPAA security regulations in particular, we must ensure the confidentiality, integrity and availability of all electronic PHI (“EPHI”) that we create, receive, maintain, or transmit. We have some flexibility to fashion our own security measures to accomplish these goals. The security regulations strongly emphasize that we must periodically conduct an accurate and thorough assessment of the potential risks and vulnerabilities of the confidentiality, integrity and availability of our EPHI and then document our response to the various security regulations on the basis of that assessment.
The privacy, security and breach notification regulations were last modified in 2013 as a result of final regulations published pursuant to the Health Information Technology Act (“HITECH”). HITECH requires, among other things, that providers, such as laboratories, notify patients of breaches of unsecured PHI, enter into new business associate agreements with existing business associates and revise many of their existing privacy policies. In addition, HITECH makes business associates directly liable to the Federal government for compliance with certain aspects of the privacy, security and breach notification regulations. As implemented in regulations, a downstream subcontractor of a business associate that creates, receives, maintains, or transmits PHI on behalf of the business associate is also itself considered a business associate. Under the regulations issued in 2013, health care providers, such as laboratories, that are subject to HIPAA as a Covered Entity are vicariously liable for violations of HIPAA based on acts or omissions of their agents, including business associates, when the agent is acting within the scope of the agency. Complying with the electronic transaction, privacy, security and breach notification rules requires significant effort and expense for virtually all entities that conduct health care transactions electronically and handle PHI.
Medical Regulated Waste
We are subject to licensing and regulation under federal, state and local laws relating to the handling and disposal of medical specimens, infectious and hazardous waste, as well as to the safety and health of laboratory employees. All our laboratories are required to operate in accordance with applicable federal and state laws and regulations relating to biohazard disposal of all facilities specimens. We use outside vendors to dispose of such specimens. Although we believe that we comply in all respects with such
| 18 |
federal, state and local laws, our failure to comply with those laws could subject us to denial of the right to conduct business, fines, criminal penalties and/or other enforcement actions.
Occupational Safety
In addition to its comprehensive regulation of safety in the workplace, the U.S. Federal Occupational Safety and Health Administration (“OSHA”) has established extensive requirements relating to workplace safety for health care employers, including clinical laboratories, whose workers may be exposed to blood-borne pathogens such as HIV and the hepatitis B virus. These regulations, among other things, require work practice controls, protective clothing and equipment, training, medical follow-up, vaccinations and other measures designed to minimize exposure to, and transmission of, blood-borne pathogens. The Federal Drug Enforcement Administration regulates the use of controlled substances in testing for drugs of abuse. We are also subject to OSHA’s requirement that employers using hazardous chemicals communicate the properties and hazards presented by those chemicals to their employees. We believe that we are in compliance with these OSHA requirements. Our failure to comply with those regulations and requirements could subject us to tort liability, civil fines, criminal penalties and/or other enforcement actions.
Other Regulation
Our business is and will continue to be subject to regulation under various state and federal environmental, safety and health laws, including the Occupational Safety and Health Act, the Resource Conservation and Recovery Act, and the Atomic Energy Act or their state law analogs. These and other laws govern our use, handling and disposal of various biological, chemical and radioactive substances used in our operations and wastes generated by our operations. We are required to possess licenses under, or are otherwise subject to federal and state regulations pertaining to, the handling and disposal of medical specimens, infectious and hazardous waste and radioactive materials.
We believe that we are in compliance with applicable environmental, safety and health laws in the United States and internationally and that our continual compliance with these laws will not have a material adverse effect on our business. All of our laboratories are operated in accordance with applicable federal and state laws and regulations relating to hazardous substances and wastes, and we use qualified third-party vendors to dispose of biological specimens and other hazardous wastes. Although we believe that we comply in all respects with such federal, state and local laws, our failure to comply with those laws could subject us to denial of the right to conduct business, civil fines, criminal penalties and/or other enforcement actions. Environmental contamination resulting from spills or disposal of hazardous substances generated by our operations, even if caused by a third-party contractor or occurring at a remote location could result in material liability.
Regulation of Diagnostic Products
The diagnostic products that are developed by our collaborators, or by us, are likely to be regulated by the FDA as medical devices. Unless an exemption applies, medical devices must receive either “510(k) clearance” or pre-market approval (“PMA”) from the FDA before marketing them in the United States. Both the 510(k) clearance and PMA processes may be costly and time consuming, but the process of obtaining PMA approval is much more costly, lengthy and uncertain. We cannot be sure that 510(k) clearance or PMA approval will ever be obtained for any product we propose to market.
The FDA decides whether a device must undergo either the 510(k) clearance or PMA approval process based upon statutory criteria. These criteria include the level of risk that the agency perceives is associated with the device and a determination whether the product is a type of device that is similar to devices that are already legally marketed. Devices deemed to pose relatively less risk are placed in either class I or II, which requires the manufacturer to submit a premarket notification requesting 510(k) clearance, unless an exemption applies. In a pre-market notification, the applicant must demonstrate that the proposed device is “substantially equivalent” in intended use and in safety and effectiveness to a legally marketed “predicate device” that is either in class I, class II, or is a “pre-amendment” class III device (i.e., one that was in commercial distribution before May 28, 1976) for which the FDA has not yet called for submission of a PMA application.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require a PMA approval. The FDA requires each manufacturer to make this determination in the first instance, but the FDA can review any such decision. If the FDA disagrees with a manufacturer’s decision not to seek a new 510(k) clearance, the agency may retroactively require the manufacturer to seek 510(k) clearance or PMA approval. The FDA also can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or PMA approval is obtained.
Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or deemed not substantially equivalent to a legally marketed class I or class II predicate device, or to a preamendment class III device, for which PMAs have not been called, are placed in class III. Such devices are required to undergo the PMA approval process in which the manufacturer must provide sufficient valid scientific evidence of the safety and effectiveness of the device. A PMA application typically requires the collection of extensive preclinical and clinical trial data and also information about the device and its
| 19 |
components regarding, among other things, device design, manufacturing and labeling. After approval of a PMA, a new PMA or PMA supplement is required in the event of a modification to the device, its labeling or its manufacturing process.
Although clinical investigations of most devices are subject to the investigational device exemption (“IDE”) requirements, clinical investigations of certain in vitro diagnostic (“IVDs”) tests are exempt from the IDE requirement provided the testing is non-invasive, does not require an invasive sampling procedure that presents a significant risk, does not introduce energy into the subject, and is not used as a diagnostic procedure without confirmation by another medically established test or procedure.
In addition, the IVD must be for use in the laboratory research phase of development and not represented as an effective IVD (i.e. labeled for Research Use Only (RUO)) or for use in product testing prior to full commercial marketing (i.e. for Investigational Use Only (IUO)). Because RUO and IUO-labeled products are exempt from most regulatory requirements it is important that they are not distributed for clinical diagnostics use. Mere placement of an RUO or IUO label on an IVD product does not render the device exempt from otherwise applicable regulatory requirements; indeed, FDA may determine that the device is intended for use in clinical diagnosis on the basis of other evidence, including how the device is marketed. FDA recommends that manufacturers assess the totality of the circumstances surrounding the distribution of their RUO and IUO labeled products to ensure that they are not engaging in practices that conflict with their labeling. The FDA expressed its intent to exercise heightened enforcement with respect to IUO and RUO devices improperly commercialized prior to receipt of FDA clearance or approval.
We have developed products that we currently distribute in the United States on a RUO basis. There can be no assurance that the FDA would agree that our distribution of these products meets the requirements for RUO distribution. Furthermore, failure by us or recipients of our RUO products to comply with the regulatory limitations on the sale and distribution of RUO devices could result in enforcement action by the FDA, including the imposition of restrictions on our distribution of these products.
Although FDA has long asserted it has jurisdiction over laboratory-developed tests, the agency has historically exercised discretion enforcement with respect to most such tests and not required laboratories that furnish these tests to comply with FDA’s regulatory requirements for medical devices. However, on July 31, 2014, the FDA issued a 60-day notice to Congress indicating that the FDA intends to issue Draft Guidance on the regulation of laboratory-developed test. In the notice, FDA indicates that it intends to end its policy of general enforcement discretion towards laboratory-developed test, and proposes the implementation of a risk-based regulatory framework. Under the proposed framework, many laboratory-developed tests would be subject to FDA’s requirements for medical devices, including registration and listing premarket review, medical device reports and quality systems regulations. The implementation of this framework would not begin until after a Final Guidance is issued and would occur over a nine year period with those tests that FDA considers to be highest risk falling under FDA’s review requirements first. The draft guidance was released in late September 2014, and a 120 – day public comment period ended February 2015.
In so far as the devices that we manufacture or distribute are subject to the premarket notification or premarket approval requirements a host of additional regulatory requirements may apply, including registration and listing the Quality System Regulation (which requires manufacturers to follow elaborate design, testing, control, documentation and other quality assurance procedures), the Medical Device Reporting regulation (which requires that manufacturers report to the FDA certain types of adverse events involving their products), labeling regulations, and the FDA’s general prohibition against promoting products for unapproved or “off label” uses. Class II devices may also be subject to special controls such as performance standards, post market surveillance, patient registries, and FDA guidelines that do not apply to class I devices. Unanticipated changes in existing regulatory requirements or adoption of new requirements could hurt our business, financial condition and results of operations.
We are subject to inspection and market surveillance by the FDA to determine compliance with regulatory requirements. If the FDA finds that we have failed to comply with applicable requirements, the agency can institute a wide variety of enforcement actions, ranging from a public warning letter to more severe sanctions such as fines, injunction, civil penalties, recall or seizure of our products, operating restrictions, partial suspension or total shutdown of production, refusal of our requests for 510(k) clearance or PMA approval of new products, withdrawal of 510(k) clearance or PMA approvals already granted, and criminal prosecution.
The FDA also has the authority to request repair, replacement or refund of the cost of any medical device manufactured or distributed by us. Our failure to comply with applicable requirements could lead to an enforcement action that may have an adverse effect on our financial condition and results of operations.
Unanticipated changes in existing regulatory requirements, our failure to comply with such requirements or adoption of new requirements could have a material adverse effect on us. We have employees to expedite the preparation and filing of documentation necessary for FDA clearances and approvals, patent issuances and licensing agreements. We cannot assure you that future clinical diagnostic products developed by us or our collaborators will not be required to be reviewed by FDA under the more expensive and time consuming pre-market approval process.
Regulation of Pharmaceutical Products
New drugs and biological drug products are subject to regulation under the Federal Food, Drug and Cosmetic Act, and biological products are also regulated under the Public Health Service Act. We believe that certain products developed by us or our collaborators will be regulated either as biological products or as new drugs. Both statutes and regulations promulgated thereunder govern, among other things, the testing, licensing, manufacturing, marketing, distributing, safety, and efficacy requirements, labeling, storage,
| 20 |
exporting, record keeping, advertising and other promotional practices involving biologics or new drugs, as the case may be. FDA review or approval or other clearances must be obtained before clinical testing, and before manufacturing and marketing, of biologics and drugs. At the FDA, the Center for Biological Evaluation and Research (“CBER”) is responsible for the regulation of biological drugs and the Center for Drug Evaluation and Research (“CDER”) is responsible for the regulation of non-biological drugs. Biological drugs are licensed and other drugs are approved before commercialization.
Any therapeutics products that we develop will require regulatory review before clinical trials, and additional regulatory approval before commercialization. New human gene medicine products as well as immune regulation products, as therapeutics, are subject to regulation by the FDA and comparable agencies in other countries. The FDA on a case-by-case basis currently reviews each protocol. In addition, the National Institutes of Health (“NIH”) is also involved in the oversight of gene therapies and the FDA has required compliance with certain NIH requirements.
Federal requirements are detailed in Title 21 of the Code of Federal Regulations (21 CFR). In addition, the FDA publishes guidance documents with respect to the development of therapeutics protocols.
Obtaining FDA approval has historically been a costly and time-consuming process. Generally, to gain FDA approval, a developer first must conduct pre-clinical studies in the laboratory evaluating product chemistry, formulation and stability and, if appropriate, in animal model systems, to gain preliminary information on safety and efficacy.
Pre-clinical safety tests must be conducted by laboratories that comply with FDA regulations governing Good Laboratory Practices (GLP). The results of those studies are submitted with information characterizing the product and its manufacturing process and controls as a part of an investigational new drug (“IND”) application, which the FDA must review and approve before human clinical trials of an investigational drug can start. The IND application includes a detailed description of the clinical investigations to be undertaken in addition to other pertinent information about the product, including descriptions of any previous human experience and the company’s future plans for studying the drug.
In order to commercialize our pharmaceutical products, we (as the sponsor) file an Investigational New Drug (“IND”) application with FDA and will be responsible for initiating and overseeing the clinical studies to demonstrate the safety and efficacy necessary to obtain FDA marketing approval of any such products. For INDs that we sponsor, we will be required to select qualified clinical sites (usually physicians affiliated with medical institutions) to supervise the administration of the investigational product. It is the sponsor’s responsibility to ensure that the investigations are conducted and monitored in accordance with FDA regulations, Good Clinical Practices (GCP) and the general investigational plan and protocols contained in the IND. This may be done using in-house trained personnel or an outside contract research organization (CRO).
Each clinical study is also reviewed approved and overseen by an Institutional Review Board (IRB). In considering an application to perform a clinical trial, IRB will consider, among other things, ethical factors and the safety of human subjects participating in the trial. Clinical trials are normally conducted in three phases, although the phases might overlap. Phase I trials, concerned primarily with the safety and tolerance of the drug, and its pharmacokinetics (or how it behaves in the body including its absorption and distribution) involve fewer than 100 subjects. Phase II trials normally involve a few hundred patients and are designed primarily to demonstrate preliminary effectiveness and the most suitable dose or exposure level for treating or diagnosing the disease or condition for which the drug is intended, although short-term side effects and risks in people whose health is impaired may also be examined. Phase III trials are expanded, adequate and well-controlled clinical trials with larger numbers of patients and are intended to gather the additional information for proper dosage and labeling of the drug. Clinical trials may take several years to complete, but the period may vary.
Certain regulations promulgated by the FDA may shorten the time periods and reduce the number of patients required to be tested in the case of certain life-threatening diseases, which lack available alternative treatments. The FDA receives reports on the progress of each phase of clinical testing, and it may require the modification, suspension or termination of clinical trials if an unwarranted risk is presented to patients. Human gene medicine products are a new category of therapeutics.
There can be no assurance regarding the length of the clinical trial period, the number of patients that the FDA will require to be enrolled in the clinical trials in order to establish the efficacy, safety, purity and/or potency of human gene medicine products, or that the clinical and other data generated will be acceptable to the FDA to support marketing approval.
After completion of clinical trials of a new product, FDA marketing approval must be obtained before the product can be sold in the United States. If the product is regulated as a new biologic, CBER requires the submission and approval of a Biologics License Application (BLA) before commercial marketing of the biologic product. If the product is classified as a new drug, we must file a New Drug Application (“NDA”) with CDER and receive approval before commercial marketing of the drug. The NDA or BLA must include results of product development, pre-clinical studies and clinical trials. The testing and approval processes require substantial time and effort and there can be no assurance that any approval will be granted on a timely basis, if at all. The median time to obtain new product approvals after submission to the FDA is approximately 12 months. If questions arise during the FDA review process, approval can take longer. Before completing its review, the FDA may seek guidance from an Advisory Panel of outside experts at a public or closed meeting. While the advice of these committees is not binding on the FDA, it is often followed. Notwithstanding the
| 21 |
submission of relevant data, the FDA might ultimately decide that the NDA or BLA does not satisfy its regulatory criteria for approval and, thus, reject the application, refuse to approve it, or require additional clinical, preclinical or chemistry studies. Even after FDA regulatory approval or licensure, a marketed drug product is subject to continual review by the FDA.
In addition, if previously unknown problems are discovered or we fail to comply with the applicable regulatory requirements, we might be restricted from marketing a product, we might be required to withdraw the product from the market, and we might possibly become subject to seizures, injunctions, voluntary recalls, or civil, monetary or criminal sanctions. In addition, the FDA may condition marketing approval on the conduct of specific post-marketing studies to further evaluate safety and effectiveness.
For commercialization of our biological or other drug products, the manufacturing processes described in our NDA or BLA must receive FDA approval and the manufacturing facility must successfully pass an inspection prior to approval or licensure of the product for sale within the United States. The pre-approval inspection assesses whether, for example, the facility complies with the FDA’s current good manufacturing practices (cGMP) regulations. These regulations elaborate testing, control, documentation, personnel, record keeping and other quality assurance procedure requirements that must be met.
Once the FDA approves our biological or other drug products for marketing, we must continue to comply with the cGMP regulations. The FDA periodically inspects biological and other drug manufacturing facilities to ensure compliance with applicable cGMP requirements. Failure to comply with the statutory and regulatory requirements subjects the manufacturer to possible legal or regulatory action, such as suspension of manufacturing, seizure of product or voluntary recall of a product.
If a developer obtains designation by the FDA of a biologic or other drug as an “orphan” for a particular use, the developer may request grants from the federal government to defray the costs of qualified testing expenses in connection with the development of such drug. Orphan drug designation is possible for drugs for rare diseases, including many genetic diseases, which means the drug is for a disease that has a prevalence of less than 200,000 patients in the United States. The first applicant who receives an orphan drug designation and who obtains approval of a marketing application for such drug acquires the exclusive marketing rights to that drug for that use for a period of seven years unless the subsequent drug can be shown to be clinically superior. Accordingly, no other company would be allowed to market an identical orphan drug with the same active ingredient for the use approved by the FDA for seven years after the approval.
Manufacturing and Research Facilities
Our integrated laboratory and scientific efforts for our three segments take place primarily at our two adjacent facilities in Farmingdale, New York. A major part of one facility is utilized by Life Science products as its global headquarters, and also for research and manufacturing with special handling capabilities and clean rooms suitable for our operations. The Life Sciences segment has centered its US logistics, reagent and kit manufacturing at its facility in Ann Arbor, Michigan, and has European logistics operations in Lausen, Switzerland. We also contract with qualified third-party contractors to manufacture our products in cases where we deem it appropriate, for example, when it is not cost-effective to produce a product ourselves or where we seek to leverage the expertise of another manufacturer in a certain area.
Employees
As of July 31, 2018, we employed 460 full-time and 43 part-time employees. Of the full-time employees, 122 were engaged in research, development, manufacturing, and marketing of research products, 294 in performing testing, marketing and billing our clinical laboratories services and 44 in finance, information technology, administrative and executive functions. Our scientific staff, including 39 individuals with post graduate degrees, possesses a wide range of experience and expertise in the areas of recombinant DNA, nucleic acid chemistry, molecular biology and immunology. We believe that we have established good relationships with our employees.
Information Systems
Information systems are used extensively in virtually all aspects of our businesses. In our clinical laboratory services business, our information systems are critical with respect to laboratory testing, billing, accounts receivable, customer service, logistics, and management of medical data. Our success depends, in part, on the continued and uninterrupted performance of our information technology systems. Computer systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters.
Moreover, despite network security measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. We have invested heavily in the upgrade of our information and telecommunications systems to improve the quality, efficiency and security of our businesses. In addition, to complement our proprietary physician connectivity solution, EnzoDirect we have a web portal version which allows physicians to receive laboratory results from any personal computer with a browser and an Internet connection.
| 22 |
Despite the precautionary measures that we have taken to prevent unanticipated problems that could affect our information technology systems, sustained or repeated system failures that interrupt our ability to process test orders, deliver test results or perform tests in a timely manner could adversely affect our reputation and result in a loss of customers and net revenues.
Quality Assurance
We consider the quality of our clinical laboratory tests to be of critical importance, and, therefore, we maintain a comprehensive quality assurance program designed to help assure accurate and timely test results. In addition to the compulsory external inspections and proficiency programs demanded by the Medicare program and other regulatory agencies, our clinical laboratory has in place systems to emphasize and monitor quality assurance.
In addition to our own internal quality control programs, our laboratory participates in numerous externally administered, blind quality surveillance programs, including on-site evaluation by the College of American Pathologies (“CAP”) proficiency testing program and the New York State survey program. The blind programs supplement all other quality assurance procedures and give our management the opportunity to review our technical and service performance from the client’s perspective.
The CAP accreditation program involves both on-site inspections of our laboratory and participation in the CAP’s proficiency testing program for all categories in which our laboratory is accredited by the CAP. The CAP is an independent nongovernmental organization of board certified pathologists, which offers an accreditation program to which laboratories can voluntarily subscribe. A laboratory’s receipt of accreditation by the CAP satisfies the Medicare requirement for participation in proficiency testing programs administered by an external source. Our clinical laboratory facilities are CAP accredited.
FORWARD - LOOKING AND CAUTIONARY STATEMENTS
This Annual Report contains “forward-looking statements” as defined in the Private Securities Litigation Reform Act of 1995. All statements other than statements of historical fact, including, without limitation, the statements under “Management’s Discussion and Analysis of Financial Condition and Results of Operations” are “forward-looking statements.” Forward-looking statements may include the words “believes,” “expects,” “plans,” “intends,” “anticipates,” “continues” or other similar expressions. These statements are based on the Company’s current expectations of future events and are subject to a number of risks and uncertainties that may cause the Company’s actual results to differ materially from those described in the forward-looking statements. Should one or more of these risks or uncertainties materialize, or should underlying assumptions prove incorrect, actual results may vary materially from those anticipated, estimated or projected. The Company assumes no obligation to revise or update any forward-looking statements for any reason, except as required by law.
The Company files annual, quarterly and current reports, proxy statements and other information with the Securities and Exchange Commission (the “SEC”). These filings are available to the public via the Internet at the SEC’s website located at http://www.sec.gov. You may also read and copy any document the Company files with the SEC at the SEC’s public reference room located at 100 F Street, N.E., Washington, D.C. 20549. For more information, please call the SEC at 1-800-SEC-0330.
The Company’s website is located at www.enzo.com. The Company makes available on its website a link to all filings that it makes with the SEC. You may request a copy of the Company’s filings with the SEC (excluding exhibits) at no cost by writing or telephoning us at the following address or telephone number:
| 23 |
Enzo Biochem, Inc.
527 Madison Ave.
New York, New York 10022
Tel: (212) 583-0100
Attn: Investor Relations
Business Risks
Our operating results may vary from period to period.
Our operating results may vary significantly from quarter to quarter and from year to year, depending on a variety of factors including:
| • | competitive conditions, including changes in third-party reimbursements; |
| • | health care reform regulations affecting providers and plan sponsors, including those stemming from the Affordable Care Act (ACA) (or its repeal, amendment or replacement); |
| • | changes in reimbursement policies from third party payers; |
| • | Foreign currency exchange rate fluctuations; |
| • | changes in tax laws, the results of tax audits or the measurement of tax uncertainties; |
| • | the timing of our research and development, sales and marketing expenses; |
| • | the introduction of new products by us or our competitors; |
| • | the success of identifying, acquiring and integrating businesses that complement our product offerings, add new technology or add presence in a market; |
| • | expenses associated with defending our intellectual property portfolio; |
| • | customer demand for our products due to changes in purchasing requirements and research needs; |
| • | general worldwide economic conditions affecting funding of research; and |
| • | seasonal fluctuations affected by weather and holiday periods. |
Consequently, results for any interim period may not necessarily be indicative of results in subsequent periods.
A significant proportion of our sales are to academic centers, funded by government grants in our major markets globally.
Governments around the world have been reviewing long term public funding of life science research in response to the problems arising from global financial pressures. As a result, the available funds for discretionary purchases from market to market have been capped or reduced based on available National budgets. Reduced grants for researchers could impact our business, in the amount, price and type of products bought and used by customers.
A significant proportion of our sales are to customers in pharmaceutical and biotech companies.
Globally, pharmaceutical companies are challenging internal budgets, and the return of investment from their R&D spend. This could impact our business, in the amount, price and type of products bought and used by customers.
Our future success will depend in part upon our ability to enhance existing products, develop and introduce new products and realize commercial acceptance of those products, in a rapidly changing technological environment.
The market for our products is characterized by rapidly changing technology, evolving industry standards and new product introductions, which may make our existing products obsolete. Our future success will depend in part upon our ability to enhance existing products, develop and introduce new products, and realize commercial acceptance of those products.
| 24 |
The development of new or enhanced products is a complex and uncertain process requiring the accurate anticipation of technological and market trends as well as precise technological execution. In addition, the successful development of new products will depend on the development of new technologies. We will be required to undertake time-consuming and costly development activities and to seek regulatory approval for these new products. We may experience difficulties that could delay or prevent the successful development, introduction and marketing of these new products. Regulatory clearance or approval of any new products may not be granted by the FDA, state-wide agency or foreign regulatory authorities on a timely basis, or at all, and the new products may not be successfully commercialized.
We may be unable to identify, acquire and integrate acquisition targets.
Our strategy envisions, if an opportunistic target is identified, future growth from acquiring and integrating similar operations and/or product or services lines. There can be no assurance that we will be able to identify suitable acquisition candidates and, once identified, to negotiate successfully their acquisition at a price or on terms and conditions favorable to us, or to integrate the operations of such acquired businesses with the existing operations. In addition, we compete for acquisition candidates with other entities, some of which have greater financial resources than ours. Failure to implement successfully our acquisition strategy would limit our potential growth.
Our inability to carry out certain of our marketing and sales plans may make it difficult for us to grow or maintain our business.
The Life Sciences product segment continues a marketing program designed to more directly service its end users, while simultaneously promoting the Enzo Life Science brand, with reference to our acquired brands. We will continue to reach out to our customers using our direct field sales force, in-house business team, the on-going enhancement of our interactive websites, continued attendance at top industry trade meetings, and publications to customers and in leading scientific journals. In addition to our direct sales, we operate worldwide through wholly-owned subsidiaries (in USA, Switzerland, Belgium, Germany, and the UK), a branch office in France and a network of third-party distributors in most other significant markets. If we are unable to successfully continue these programs, we may be unable to grow and our business could suffer.
We face significant competition, which could cause us to decrease the prices for our products or services or render our products uneconomical or obsolete, any of which could reduce our revenues and limit our growth.
Our competitors in the biotechnology industry in the United States and abroad are numerous and include major pharmaceutical, energy, food and chemical companies, as well as specialized genetic engineering firms. Many of our large competitors have substantially greater resources than us and have the capability of developing products which compete directly with our products. Many of these companies are performing research in the same areas as we are. The markets for our products are also subject to competitive risks because markets are highly price competitive. Our competitors have competed in the past by lowering prices on certain products.
The clinical laboratory services business is highly fragmented and intensely competitive, and we compete with numerous national and local companies. Some of these entities are larger than we are and have greater resources than we do. We compete primarily on the basis of the quality of our testing, reporting and information services, our reputation in the medical community, the pricing of our services and our ability to employ qualified professionals.
These competitive conditions could, among other things:
| • | Require us to reduce our prices to retain market share; |
| • | Require us to increase our marketing efforts which could reduce our profit margins; |
| • | Increase our cost of labor to attract qualified personnel; |
| • | Render our biotechnology products uneconomical or obsolete or; |
| • | Reduce our revenue. |
Ethical, legal and social concerns surrounding the use of genetic information could reduce demand for our products.
Genetic testing has raised ethical issues regarding privacy and the appropriate uses of the resulting information. For these reasons, governmental authorities may call for limits on or regulation of the use of genetic testing or prohibit testing for genetic predisposition to certain conditions, particularly for those that have no known cure. Similarly, such concerns may lead individuals to refuse to use genetics tests even if permissible. Any of these scenarios could reduce the potential markets for our molecular diagnostic products, which could have a material adverse effect on our business, financial condition and results of operations.
| 25 |
We depend on distributors and contract manufacturers and suppliers for materials that could impair our ability to manufacture or
distribute our products.
We manufacture and distribute our own brand products and the products of third party manufacturers and suppliers. Distributors also sell our branded products. To the extent we are unable to maintain or replace a distributor in a reasonable time period, or on commercially reasonable terms, if at all, our operations could be disrupted.
Outside distributors, suppliers and contract manufacturers provide key finished goods, components and raw materials used in the sale and manufacture of our products. Although we believe that alternative sources for components and raw materials are available, any supply interruption in a limited or sole source component or raw material would harm our ability to manufacture our products until a new source of supply is identified and qualified. In addition, an uncorrected defect or supplier’s variation in a component or raw material, either unknown to us or incompatible with our manufacturing process, could harm our ability to manufacture products. We might not be able to find a sufficient alternative supplier in a reasonable time period, or on commercially reasonable terms, if at all. If we fail to obtain a supplier for the components of our products, our operations could be disrupted.
We use hazardous materials in our business. Any claims relating to improper handling, storage or disposal of these materials could be costly and time-consuming.
Our manufacturing, clinical laboratory and research and development processes involve the storage, use and disposal of hazardous substances, including hazardous chemicals, biological hazardous materials and radioactive compounds. We are subject to governmental regulations governing the use, manufacture, storage, handling and disposal of materials and waste products. Although we believe that our safety and environmental management practices and procedures for handling and disposing of these hazardous materials are in accordance with good industry practice and comply with applicable laws, permits, licenses and regulations, the risk of accidental environmental or human contamination or injury from the release or exposure of hazardous materials cannot be completely eliminated. In the event of an accident, we could be held liable for any damages that result, including environmental clean-up or decontamination costs, and any such liability could exceed the limits of, or fall outside the coverage of, our insurance.
We may not be able to maintain insurance on acceptable terms, or at all. We could be required to incur significant costs to comply with current or future environmental and public and workplace safety and health laws and regulations.
We are required to expend significant resources for research and development for our products in development and these products may not be developed successfully. Failure to successfully develop these products may prevent us from earning a return on our research and development expenditures.
The products we are developing are at various stages of development and clinical evaluations and may require further technical development and investment to determine whether commercial application is practicable. There can be no assurance that our efforts will result in products with valuable commercial applications. Our cash requirements may vary materially from current estimates because of results of our research and development programs, competitive and technological advances and other factors. In any event, we will require substantial funds to conduct development activities and pre-clinical and clinical trials, apply for regulatory approvals and commercialize products, if any, that are developed.
We do not have any commitments or arrangements to obtain any additional financing and there is no assurance that required financing will be available to us on acceptable terms, if at all. Even if we spend substantial amounts on research and development, our potential products may not be developed successfully.
If our product candidates on which we have expended significant amounts for research and development are not commercialized, we will not earn a return on our research and development expenditures, which may harm our business.
We rely on network and information systems and other technology whose failure or misuse could cause a disruption of services or loss or improper disclosure of personal data, business information, including intellectual property, or other confidential information, resulting in increased costs, loss of revenue or other harm to our business.
Network and information systems and other technologies, including those related to the Company’s network management, are important to its business activities. The Company also relies on third party providers for certain technology and “cloud-based” systems and services that support a variety of business operations. Network and information systems-related events affecting the Company’s systems, or those of third parties upon which the Company’s business relies, such as computer compromises, cyber threats and attacks, computer viruses, worms or other destructive or disruptive software, process breakdowns, denial of service attacks, malicious social engineering or other malicious activities, or any combination of the foregoing, as well as power outages, equipment failure, natural disasters (including extreme weather), terrorist activities, war, human or technological error or malfeasance that may affect such systems, could result in disruption of the Company’s business and/or loss, corruption or improper disclosure of personal data, business
| 26 |
information, including intellectual property, or other confidential information. In addition, any design or manufacturing defects in, or the improper implementation of, hardware or software applications the Company develops or procures from third parties could
unexpectedly compromise information security. In recent years, there has been a rise in the number of cyber-attacks on companies’ network and information systems, and such attacks have become more sophisticated, targeted and difficult to detect and prevent against. As a result, the risks associated with such an event continue to increase, particularly as the Company’s digital businesses expand. While the Company has developed and implemented security measures and internal controls that are designed to protect personal data, business information, including intellectual property, and other confidential information, to prevent data loss, and to prevent or detect security breaches, such security measures cannot provide absolute security and may not be successful in preventing these events from occurring, particularly given that techniques used to access, disable or degrade service, or sabotage systems change frequently, and any network and information systems-related events could require the Company to expend significant resources to remedy such event. Moreover, the development and maintenance of these measures is costly and requires ongoing monitoring and updating as technologies change and efforts to overcome security measures become more sophisticated. While the Company maintains cyber risk insurance, this insurance may not be sufficient to cover all losses from any future breaches of our systems.
A significant failure, compromise, breach or interruption of the Company’s systems, or those of third parties upon which its business relies, could result in a disruption of its operations, customer, audience or advertiser dissatisfaction, damage to its reputation or brands, regulatory investigations and enforcement actions, lawsuits, remediation costs, a loss of customers, advertisers or revenues and other financial losses. If any such failure, interruption or similar event results in the improper disclosure of information maintained in the Company’s information systems and networks or those of its vendors, including financial, personal, credit card, confidential and proprietary information relating to personnel, customers, vendors and the Company’s business, including its intellectual property, the Company could also be subject to liability under relevant contractual obligations and laws and regulations protecting personal data and privacy. In addition, media or other reports of perceived security vulnerabilities to our systems or those of third parties upon which the Company’s business relies, even if nothing has actually been attempted or occurred, could also adversely impact our brand and reputation and materially affect our business.
Risks relating to our Intellectual Property and Regulatory Approval
Protecting our proprietary rights is difficult and costly. If we fail to adequately protect or enforce our proprietary rights, we could lose potential revenue from licensing and royalties.
Our potential revenue and success depends in large part on our ability to obtain, maintain and enforce our patents. Our ability to commercialize any product successfully will largely depend on our ability to obtain and maintain patents of sufficient scope to prevent third parties from developing similar or competitive products. In the absence of patent protection, competitors may impact our business by developing and marketing substantially equivalent products and technology.
Patent disputes are frequent and can preclude the commercialization of products. We have in the past been, are currently, and may in the future be, involved in material patent litigation, such as the matters discussed under “Part I - Item 3. Legal Proceedings” in this report. Patent protection litigation is time-consuming and we have incurred and anticipate continuing to incur significant legal costs. In addition, an adverse decision could force us to either obtain third-party licenses at a material cost or cease using the technology or product in dispute.
We have filed applications for United States and foreign patents covering certain aspects of our technology, but there is no assurance that pending patents will issue or as to the degree of protection which any issued patent might afford.
Lawsuits, including patent infringements, in the biotechnology industry are not uncommon. If we become involved in any significant litigation, we would suffer as a result of the diversion of our management’s attention, the expense of litigation and any judgments against us.
In addition to intellectual property litigation for infringement, other substantial, complex or extended litigation could result in large expenditures by us and distraction of our management. Patent litigation is time-consuming and costly in its own right and could subject us to significant liabilities to third parties. In addition, an adverse decision could force us to either obtain third-party licenses at a material cost or cease using the technology or product in dispute. In addition, lawsuits by employees, stockholders, collaborators or distributors could be very costly and substantially disrupt our business. Disputes from time to time with companies or individuals are not uncommon in the biotechnology industry, and we cannot assure you that we will always be able to resolve them out of court.
We also utilize certain unpatented proprietary technology and no assurance can be given that others will not independently develop substantially equivalent proprietary technology, that such proprietary technology will not be disclosed or that we can meaningfully protect our rights to such proprietary technology.
| 27 |
Our business is subject to governmental laws and regulations. Changes in the way the FDA regulates the reagents, and other consumables we use when developing, validating, and performing our tests could result in delay or additional expense in bringing our tests to market or performing such tests for our customers. We may be unable to obtain or maintain regulatory approvals for our products, which could reduce our revenue or prevent us from earning a return on our research and development expenditures.
Our research, preclinical development, clinical trials, product manufacturing and marketing are subject to regulation by the FDA and similar health authorities in foreign countries. FDA approval is required for our products, as well as the manufacturing processes and facilities, if any, used to produce our products that may be sold in the United States. The process of obtaining approvals from the FDA is costly, time consuming and often subject to unanticipated delays. Even if regulatory approval is granted, such approval may include significant limitations on indicated uses for which any products could be marketed. Further, even if such regulatory approvals are obtained, a marketed product and its manufacturer are subject to continued review, and later discovery of previously unknown problems may result in restrictions on such product or manufacturer, including withdrawal of the product from the market.
New government regulations in the United States or foreign countries also may be established that could delay or prevent regulatory approval of our products under development. Further, because gene therapy is a relatively new technology and has not been extensively tested in humans, the regulatory requirements governing gene therapy products are uncertain and may be subject to substantial further review by various regulatory authorities in the United States and abroad. This uncertainty may result in extensive delays in initiating clinical trials and in the regulatory approval process. Our failure to obtain regulatory approval of our proposed products, processes or facilities could have a material adverse effect on our business, financial condition and results of operations. The proposed products under development may also be subject to certain other federal, state and local government regulations, including, but not limited to, the Federal Food, Drug and Cosmetic Act, the Environmental Protection Act, and Occupational Safety and Health Act, and state, local and foreign counterparts to certain of such acts.
In November 2013 the FDA issued a Guidance document entitled “Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only,” or the RUO Guidance, which highlights the FDA’s interpretation that distribution of RUO products with any labeling, advertising or promotion that suggests that clinical laboratories can validate the test through their own procedures and subsequently offer it for clinical diagnostic use as an LDT is in conflict with RUO status. The RUO Guidance further articulates the FDA’s position that any assistance offered in performing clinical validation or verification, or similar specialized technical support, to clinical laboratories, is in conflict with RUO status. More recently, on October 3, 2014, the FDA announced the availability of a draft guidance entitled “Framework for Regulatory Oversight of Laboratory-Developed Tests,” a risk-based oversight framework for LDTs. If the draft guidance is finalized as presently written, such oversight framework includes a premarket review for higher-risk LDTs, such as those that have the same intended use as an FDA-approved or cleared companion diagnostic currently on the market, as well as other high risk and moderate risk LDTs over time. As a result of the draft guidance, we may be required to seek clearance or approval to offer our tests for clinical use earlier than we otherwise might have done. If we engage in any activities that are in conflict with the RUO status held by some of the tests that we sell or intend to sell, we may be subject to immediate, severe and broad FDA enforcement action that would adversely affect our ability to continue operations. Accordingly, if the FDA finds that we are distributing our RUO products in a manner that is inconsistent with its guidance, we may be forced to stop distribution of our RUO tests until we are in compliance, which, would reduce our revenue, increase our costs and adversely affect our business, prospects, results of operations and financial condition.
We cannot be sure that we can obtain necessary regulatory approvals on a timely basis, if at all, for any of the products we are developing or manufacturing or that we can maintain necessary regulatory approvals for our existing products, and all of the following could have a material adverse effect on our business:
| • | significant delays in obtaining or failing to obtain required approvals; |
| • | loss of, or changes to, previously obtained approvals; |
| • | failure to comply with existing or future regulatory requirements and; |
| • | changes to manufacturing processes, manufacturing process standards or Good Manufacturing Practices following approval or changing interpretations of these factors. |
Adverse perception and increased regulatory scrutiny of gene medicine and genetic research might limit our ability to conduct our business.
Ethical, social and legal concerns about gene medicine, genetic testing and genetic research could result in additional regulations restricting or prohibiting the technologies we or our collaborators may use. Recently, gene medicine studies have come under increasing scrutiny, which has delayed on-going and could delay future clinical trials and regulatory approvals. Federal and state
| 28 |
agencies, congressional committees and foreign governments have expressed interest in further regulating biotechnology. More restrictive regulations or claims that our products are unsafe or pose a hazard could prevent us from commercializing any products.
Financial Risks
With the exception of 2016, we have experienced significant losses in our previous five fiscal years and quarter to quarter over such periods and our losses have resulted in the use of cash in operations. If such losses and cash uses continue, the value of your investment could decline significantly.
Although for fiscal year 2016, we reported net income of $45.3 million, we incurred net losses of $10.3 million and $2.5 million for the fiscal years ended July 31, 2018 and 2017 respectively. If our revenues do not increase, or if our operating expenses exceed expectations or cannot be reduced, we will continue to suffer substantial losses and use cash in operations which could have an adverse effect on our business and adversely affect your investment in our Company.
We may need additional capital to fund growth, which may not be available on acceptable terms or at all, and could result in our business plan being limited and our business being harmed.
Our ability to increase revenue and improve profitability and liquidity will depend in part on our ability to grow our products business with higher margin products and increase our market share and continue to grow the Laboratory Services business with new tests with higher reimbursements and increase our service volume which may require significant additional capital that may not be available to us. We may need additional financing due to future developments, changes in our business plan or failure of our current business plan to succeed, which could result from increased marketing, distribution or research and development costs. Our actual funding requirements could vary materially from our current estimates. If additional financing is needed, we may not be able to raise sufficient funds on favourable terms or at all. If we issue common stock or securities convertible into common stock in the future, such issuance will result in the then-existing stockholders sustaining dilution to their relative proportion of our outstanding equity. If we fail to obtain any necessary financing on a timely basis, then our ability to execute our current business plan may be limited, and our business, liquidity and financial condition could be harmed.
We may incur impairment charges on our goodwill and intangibles which would reduce our earnings.
We are subject to Statement of Financial Accounting Standards ASC 350, “Intangibles - Goodwill and Other (“ASC 350”) which requires that goodwill and other intangible assets that have an indefinite life be tested at least annually for impairment. Goodwill and other intangible assets with indefinite lives must also be tested for impairment between the annual tests if a triggering event occurs that would likely reduce the fair value of the asset below its carrying amount.
As of July 31, 2018 and 2017, goodwill and intangible assets represented approximately 9% and 10%, respectively, of our total assets. If we determine that there has been impairment, our financial results for the relevant period would be reduced by the amount of the impairment, net of tax effects, if any. The Company has no intangible assets with indefinite lives.
Risks relating to our Clinical Laboratory Services segment
Our clinical laboratory services business is subject to extensive government regulation and our loss of any required certifications or licenses could require us to cease operating this part of our business, which would reduce our revenue and injure our reputation.
The clinical laboratory industry is subject to significant governmental regulation at the Federal, state and local levels. Under the Clinical Laboratory Improvement Act of 1967 and the Clinical Laboratory Improvement Amendments of 1988 (collectively, as amended, “CLIA”) virtually all clinical laboratories, including ours, must be certified by the Federal government. Many clinical laboratories also must meet other governmental standards, undergo proficiency testing and are subject to inspection. Certifications or licenses are also required by various state and local laws. The failure of our clinical laboratory to obtain or maintain such certifications or licenses under these laws could interrupt our ability to operate our clinical laboratory business and injure our reputation. The Protecting Access to Medicare Act (“PAMA”) of 2014 includes a substantial new payment system for clinical laboratory tests under the Clinical Laboratory Fee Schedule, or CLFS and has impacted the clinical laboratory testing industry. Key parts of this legislation include provisions that provide for the establishment of an advisory panel and a market-based process to rebase the clinical laboratory fee schedule, developing a new fee schedule and limiting reductions in that fee schedule. If this process does not recognize the value that clinical laboratory testing brings to the healthcare system, our business can be materially adversely impacted.
| 29 |
Reimbursements from third-party payers including managed care organizations and Medicare, upon which our clinical laboratory business is dependent, are subject to inconsistent rates and coverage and legislative reform that are beyond our control. This inconsistency and any reform that decreases coverage and rates could reduce our earnings and harm our business.
Our clinical laboratory services business is primarily dependent upon reimbursement from third-party payers, such as Medicaid, Medicare (which principally serves patients 65 and older) and commercial insurers. We are subject to variances in reimbursement rates among different third-party payers, as well as constant renegotiation of those reimbursement rates. Government and non-government payers have in the past sought, and continue to seek, to reduce and limit utilization and reimbursement of healthcare services, including the areas of clinical and genetic testing. We also are subject to audit by Medicare and the commercial insurers, which can result in the return of payments made to us under these programs. These variances in reimbursement rates and audit results could reduce our margins and thus our earnings.
The health care industry continues to undergo significant change as third-party payers’ increase their efforts to control the cost, utilization and delivery of health care services. In an effort to address the problem of increasing health care costs, legislation has been proposed or enacted at both the Federal and state levels to regulate health care delivery in general and clinical laboratories in particular. Some of the proposals include managed competition, global budgeting and price controls. Changes that decrease reimbursement rates or coverage, or increase administrative burdens on billing third-party payers could reduce our revenues and increase our expenses.
Since each payer makes its own decision as to whether to establish a policy or enter into a contract to cover our tests, as well as the amount it will reimburse for a test, seeking these approvals is a time-consuming and costly process. In addition, the determination by a payer to cover and the amount it will reimburse for our tests will likely be made on an indication by indication basis. To date, we have obtained policy-level reimbursement approval or contractual reimbursement for some indications for our test from a small number of commercial third-party payers, and have not obtained coverage from Medicare or any state Medicaid program. Further, we believe that establishing adequate reimbursement from Medicare is an important factor in gaining adoption from healthcare providers. Our claims for reimbursement from commercial payers may be denied upon submission, and we must appeal the claims. The appeals process is time consuming and expensive, and may not result in payment. In cases where there is not a contracted rate for reimbursement, there is typically a greater co-insurance or co-payment requirement from the patient which may result in further delay or decreased likelihood of collection.
We expect to continue to focus substantial resources on increasing adoption of, and coverage and reimbursement for, our current tests and any future tests we may develop. We believe it may take several years to achieve coverage and adequate contracted reimbursement with a majority of third-party payers. However, we cannot predict whether, under what circumstances, or at what payment levels payers will reimburse for our tests. If we fail to establish and maintain broad adoption of, and coverage and reimbursement for, our tests, our ability to generate revenue could be harmed and our future prospects and our business could suffer.
U.S. healthcare reform legislation may result in significant change and our business could be adversely impacted if we fail to adapt.
Government oversight of and attention to the healthcare industry in the United States is significant and increasing. Under the Patient Protections and affordable Care Act, expansion in the pool of covered lives may expand the market for clinical diagnostic testing while at the same time, various policies aimed at reducing costs or bundling care may reduce the rates paid for such services’ the net impact of these factors on the market for our tests is not clear. In April 2014, Congress passed the Protecting Access to Medicare Act of 2014, which include substantial changes to the way in which clinical laboratory services will be paid under Medicare. Beginning on January 1, 2018, Medicare payments for clinical laboratory services are paid based upon private payer rates reported by clinical laboratories across the US replacing the current system, which is based upon fee schedules derived from historical charges for test from approximately 30 years ago. While we believe that the new rates will have minimal impact on our business, it is unclear whether and to what extent the new rates will affect overall pricing and reimbursement for clinical laboratory testing services.
The Patient Protection and Affordable Care Act also imposes an excise tax on the seller for the sale of certain medical devices in the United States, including those purchased and used by laboratories, beginning in 2013. The legislation also establishes the Independent Payment Advisory Board, which will be responsible, beginning in 2014, annually to submit proposals aimed at reducing Medicare cost growth while preserving quality. If the projected growth in per capita Medicare costs exceeds a specified target level, the IPAB must submit proposals to reduce or eliminate the difference. For calendar years 2015 through 2019, the target growth rate is the projected average of the increases in the Consumer Price Index and the medical care expenditure category of the Consumer Price Index; for 2020 and thereafter, the target growth rate is the rate of increase in gross domestic product per capita plus one percentage point. If it is necessary for the IPAB to submit proposals, they will automatically be implemented unless Congress enacts alternative proposals that achieve the same savings targets.
| 30 |
Further, the legislation calls for a Center for Medicare and Medicaid Innovation that will examine alternative payment methodologies and conduct demonstration programs. The legislation provides for extensive health insurance reforms, including the elimination of pre-existing condition exclusions and other limitations on coverage, fixed percentages on medical loss ratios, expansion in Medicaid and other programs, employer mandates, individual mandates, creation of state and regional health insurance exchanges, and tax subsidies for individuals to help cover the cost of individual insurance coverage. The legislation also permits the establishment of accountable care organizations, a new healthcare delivery model. While the ultimate impact of the legislation on the healthcare industry is unknown, it is likely to be extensive and may result in significant change. Our failure to adapt to these changes could have a material adverse effect on our business.
Changes in provider mix, including continued growth in capitated managed-cost health care and changes in certain third party provider agreements could have a material adverse impact on the Company’s net revenues and profitability.
Certain third party provider companies have adopted national and regional programs which include multiple managed-care reimbursement models. If the Company is unable to participate in these programs or if the Company would lose a material contract, it could have a material adverse impact on the Company’s net revenues and profitability.
The number of individuals covered under managed care contracts or other similar arrangements has grown over the past several years and may continue to grow in the future. In addition, Medicare and other government healthcare programs may continue to shift to managed care. Entities providing managed care coverage have reduced payments for medical services, including clinical laboratory services, in numerous ways such as entering into arrangements under which payments to a service provider are capitated, limiting testing to specified procedures, denying payment for services performed without prior authorization and refusing to increase fees for specified services. These trends reduce our revenues and limit our ability to pass cost increases to our customers. Also, if these or other managed care organizations do not select us as a participating provider, we may lose some or all of that business, which could have an adverse effect on our business, financial condition and results of operations.
Because of competitive pressures, impacts of the economy on patient visits to our customer physician locations and the complexity and expense of the billing process in our clinical laboratory services business, we must obtain new customers while maintaining existing customers to grow our business.
Intense competition in the clinical laboratory business, increasing administrative burdens upon the reimbursement process, reduced patient traffic, and reduced coverage and payments by insurers make it necessary for us to increase our volume of laboratory services. To do so, we must obtain new customers while retaining existing customers.
Our failure to attract new customers or the loss of existing customers or a reduction in business from those customers could significantly reduce our revenues and impede our ability to grow.
Compliance with Medicare administrative policies, including those pertaining to certain automated blood chemistry profiles, may reduce the reimbursements we receive.
Containment of health care costs, including reimbursement for clinical laboratory services, has been a focus of on-going governmental activity. Clinical laboratories must bill Medicare directly for the services provided to Medicare beneficiaries and may only collect the amounts permitted under this fee schedule. Reimbursement to clinical laboratories under the Medicare Fee Schedule has been steadily declining since its inception. Because a significant portion of our costs is fixed, these Medicare reimbursement reductions and changes have a direct adverse effect on our net earnings and cash flows.
The development of new, more cost-effective tests that can be performed by our customers or by patients, and the continued internalization of testing by hospitals or physicians, could negatively impact our testing volume and revenues.
The diagnostic industry is faced with changing technology and new product introductions, including technology that enables more convenient or cost-effective testing. Some of our competitors also may offer testing to be performed outside of a commercial clinical laboratory, such as point-of-care testing that can be performed by physicians in their offices; complex testing that can be performed by hospitals in their own laboratories; and home testing that can be carried out without requiring the services of outside providers.
Advances in technology also may lead to the need for less frequent testing. Further, diagnostic tests approved or cleared by the FDA for home use are automatically deemed to be “waived” tests under CLIA and may be performed by patients in their homes; test kit manufacturers could seek to increase sales to patients of such test kits. Development of such technology and its use by our customers would reduce the demand for our laboratory-based testing services and negatively impact our revenues.
Our business could be harmed from the loss or suspension of a license or imposition of a fine or penalties under, or future changes in, or changing interpretations of, CLIA or state laboratory licensing laws to which we are subject.
| 31 |
The clinical laboratory testing industry is subject to extensive federal and state regulation, and many of these statutes and regulations have not been interpreted by the courts. The Clinical Laboratory Improvement Amendments of 1988, or CLIA, are federal regulatory standards that apply to virtually all clinical laboratories (regardless of the location, size or type of laboratory), including those operated by physicians in their offices, by requiring that they be certified by the federal government or by a federally approved accreditation agency. CLIA does not pre-empt state law, which in some cases may be more stringent than federal law and require additional personnel qualifications, quality control, record maintenance and proficiency testing. The sanction for failure to comply with CLIA and state requirements may be suspension, revocation or limitation of a laboratory’s CLIA certificate, which is necessary to conduct business, as well as significant fines and/or criminal penalties. Several states have similar laws and we may be subject to similar penalties.
We cannot assure that applicable statutes and regulations will not be interpreted or applied by a prosecutorial, regulatory or judicial authority in a manner that would adversely affect our business. Potential sanctions for violation of these statutes and regulations include significant fines and the suspension or loss of various licenses, certificates and authorizations, which could have a material adverse effect on our business. In addition, compliance with future legislation could impose additional requirements on us, which may be costly.
Regulations requiring the use of “standard transactions” for healthcare services may negatively impact our profitability and cash flows.
The administrative simplification provisions of the Health Insurance Portability and Accountability Act of 1996 and its implementing regulations, or HIPAA, were designed to improve the efficiency and effectiveness of the healthcare system by facilitating the electronic exchange of information in certain financial and administrative transactions while protecting the privacy and security of the information exchanged. The administrative simplification provisions address standards for electronic transactions, security regulations and privacy regulations.
The HIPAA transaction standards are complex, and subject to differences in interpretation by payers. For instance, some payers may interpret the standards to require us to provide certain types of information, including demographic information not usually provided to us by physicians. While most of our transactions are submitted and/or received in ANSI standard format, inconsistent application of transaction standards by some remaining payers or our inability to obtain certain billing information not usually provided to us by physicians could increase our costs and the complexity of billing. In addition, new requirements for additional standard transactions, such as claims attachments, could prove technically difficult, time-consuming or expensive to implement. We are working closely with our payers to establish acceptable protocols for claims submissions and with our industry trade association and an industry coalition to present issues and problems as they arise to the appropriate regulators and standards setting organizations.
Compliance with the HIPAA security, privacy and breach notification regulations and privacy regulations may increase our costs.
The HIPAA privacy and security and breach notification regulations establish comprehensive federal standards with respect to the uses and disclosures PHI by Covered Entities. These regulations were recently amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, or HITECH, to, among other things directly apply to business associates (i.e., individuals or entities who create, receive, maintain or transmit PHI on behalf of a Covered Entity in performing functions or activities regulated by HIPAA or who perform certain services, other than treatment, on behalf of Covered Entities and receive PHI in order to perform such services) with regard to certain requirements. The regulations also specify that business associates include subcontractors that create, receive, maintain or transmit PHI on behalf of a business associate. The regulations establish a complex regulatory framework on a variety of subjects, including:
| ▪ | the circumstances under which uses and disclosures of PHI are permitted or required without a specific authorization by the patient, including but not limited to treatment purposes, activities to obtain payments for our services, and our healthcare operations activities; | |
| ▪ | a patient’s rights to access, amend and receive an accounting of certain disclosures of PHI; | |
| ▪ | requirements to notify individuals if there is a breach of their PHI; | |
| ▪ | the requirements for business associates and the terms of business associate agreements; | |
| ▪ | the content of notices of privacy practices for protected health information and; | |
| ▪ | administrative, technical and physical safeguards required of entities that use or receive PHI. |
| 32 |
We have implemented practices to meet the requirements of the HIPAA privacy, security and breach notification regulations, and updated these practices to comply with HITECH. HIPAA establishes a “floor” and does not supersede state laws that are more stringent. Therefore, we are required to comply with federal privacy security and breach notification regulations and varying state privacy, security and breach notification laws and regulations. In addition, for healthcare data transfers from other countries relating to citizens of those countries, we must comply with the laws of those other countries. The federal privacy regulations restrict our ability to use or disclose patient-identifiable laboratory data, without patient authorization, for purposes other than payment, treatment, healthcare operations and certain other specified disclosures such as public health and governmental oversight of the health care industry. The privacy, security and breach notification regulations provide for significant fines and other penalties for wrongful use or disclosure of PHI, including potential civil and criminal fines and penalties. Although the HIPAA statute and regulations do not expressly provide for a private right of damages, we also could incur damages under state laws to private parties for the wrongful use or disclosure of confidential health information or other private personal information.
Compliance with all of the HIPAA regulations, including standard transactions, requires on-going resources from all healthcare organizations, not just clinical laboratories. While we believe our total costs to comply with HIPAA will not be material to our operations or cash flows, new standard transactions and additional customer requirements resulting from different interpretations of the current regulations could impose additional costs on us.
FDA regulation of laboratory-developed tests, analyte specific reagents, or genetic testing could lead to increased costs and delays in introducing new genetic tests.
The FDA has regulatory responsibility over, among other areas, instruments, test kits, reagents and other devices used by clinical laboratories to perform diagnostic testing in the U.S. A number of tests we develop internally are offered as lab developed tests (LDTs). The FDA has claimed regulatory authority over all LDTs, but has stated that it exercised enforcement discretion with regard to most LDTs performed by high complexity CLIA-certified laboratories. The FDA has published a “Discussion Document” that provides the FDA’s views on legislation to govern LDTs. New legislation could significantly impact the clinical laboratory testing business, including by increasing or modifying the regulation of LDTs, hindering our ability to develop and market new services, causing an increase in the cost of our services, delaying our ability to introduce new tests or hindering our ability to perform testing.
We are subject to federal and state healthcare fraud and abuse and other laws and regulations and could face substantial penalties if we are unable to fully comply with such laws.
As a provider of clinical laboratory testing services, we are subject to extensive and frequently changing federal, state and local laws and regulations governing various aspects of our business. For example, we are subject to healthcare fraud and abuse regulation and enforcement by both the federal government and the states in which we conduct our business. These healthcare laws and regulations include, for example:
| ▪ | the federal Anti-Kickback Law, which constrains our marketing practices, educational programs, pricing policies, and relationships with healthcare providers or other entities, including third-party laboratories, by prohibiting, among other things, persons or entities from soliciting, receiving, offering or providing remuneration, directly or indirectly, in return for or to induce either the referral of an individual for, or the purchase, lease order or recommendation of, any good, facility, item or services for which payment may be made, in whole or in part, under a federal healthcare program such as the Medicare and Medicaid programs; | |
| ▪ | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payers that are false or fraudulent, and which may apply to entities like us to the extent that our interactions with customers may affect their billing or coding practices; | |
| ▪ | the federal Health Insurance Portability and Accountability Act of 1996, as amended, or HIPAA, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information, and also established federal crimes for knowingly and willfully executing a scheme to defraud any healthcare benefit program or making false statements in connection with the delivery of or payment for healthcare benefits, items or services, and which imposed certain requirements relating to privacy, security, and transmission of individually identifiable health information; | |
| ▪ | the federal physician self-referral law, commonly known as the Stark Law, which prohibits a physician from making a referral to an entity for certain designated health services reimbursed by Medicare or Medicaid if the physician (or a member of the physician’s family) has a financial relationship with the entity, and which also prohibits the submission of any claims for reimbursement for designated health services furnished pursuant to a prohibited referral; | |
| ▪ | the federal Physician Payment Sunshine Act, and its implementing regulations, which requires manufacturers of certain drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s |
| 33 |
| Health Insurance Program (with certain exceptions) to report annually to the United States Department of Health and Human Services information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; | ||
| ▪ | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and | |
| ▪ | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payer, including commercial insurers and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, with differing effects. |
We are unable to predict what additional federal or state legislation or regulatory initiatives may be enacted in the future regarding our business or the healthcare industry in general, or what effect such legislation or regulations may have on us. Federal or state governments may impose additional restrictions or adopt interpretations of existing laws that could have an adverse effect on us.
We incur significant costs in complying with these laws and regulations. Because of the breadth of these laws and the narrowness of available statutory and regulatory exemptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations, or our sales techniques or product placement strategies, are found to be in violation of, or to encourage or assist the violation by third parties of, any of the laws described above or any other governmental regulations that apply to us, or if we fail to maintain, renew or obtain necessary permits, licenses and approvals related to our in-house laboratory, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from the Medicare and Medicaid programs, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, suspension or revocation of certifications or licenses that are required to operate our business, injunctions and other associated remedies, the curtailment or restructuring of our operations, denial or withdrawal of product clearances, or private “qui tam” actions brought by individual whistleblowers in the name of the government, any of which could have an adverse effect on our business. If we or others determine that any of our existing customer relationships do not comply with applicable laws and regulations, either due to changes in such laws and regulations or evolving interpretations of such laws and regulations, we may be required to renegotiate or terminate such relationships. Any penalties, damages, fines, exclusions, curtailment or restructuring of our operations could adversely affect our ability to operate our business and our financial results. The risk of our being found in violation of these laws is increased by the fact that many of these laws are broad and their provisions are open to a variety of interpretations. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
Other risks relating to our business
If we fail to maintain or monitor our information systems our businesses could be adversely affected.
We depend on information systems throughout our Company to control our manufacturing, inventory, distribution and website and the clinical laboratory services processes for: processing specimens, managing inventory, processing test results and submitting claims, collecting from insurers and patients, responding to inquiries, contributing to our overall internal control processes, maintaining records of our property, plant and equipment, and recording and paying amounts due vendors and other creditors. If we were to experience a prolonged disruption in our information systems that involve interactions with customers and suppliers, it could result in the loss of sales and customers and/or increased costs, which could adversely affect our business.
Cyber security risks and the failure to maintain the confidentiality, integrity, and availability of our computer hardware, software, and Internet applications and related tools and functions could result in damage to the Company’s reputation and/or subject the Company to costs, fines, or lawsuits.
The integrity and protection of our own data, and that of its customers and employees, is critical to the Company’s business. The regulatory environment governing information, security and privacy laws is increasingly demanding and continues to evolve. Maintaining compliance with applicable security and privacy regulations may increase the Company’s operating costs and/or adversely impact the Company’s ability to market its products and services to customers. Although the Company’s computer and communications hardware is protected through physical and software safeguards, it is still vulnerable to fire, storm, flood, power loss, earthquakes, telecommunications failures, physical or software break-ins, software viruses, and similar events. These events could lead to the unauthorized access, disclosure and use of non-public information. The techniques used by criminal elements to attack computer systems are sophisticated, change frequently and may originate from less regulated and remote areas of the world. As a result, the Company may not be able to address these techniques proactively or implement adequate preventative measures. If the Company’s computer systems are compromised, it could be subject to fines, damages, litigation, and enforcement actions, customers
| 34 |
could curtail or cease using its applications, and the Company could lose trade secrets, the occurrence of which could harm its business.
If we fail to attract and retain key personnel, including our senior management, our business could be adversely affected.
Most of our products and services are highly technical in nature. In general, only highly qualified and trained scientists and technician personnel have the necessary skills to develop proprietary technological products and market our products, support our research and development programs and provide our clinical laboratory services.
In addition, some of our manufacturing, quality control, safety and compliance, information technology and e-commerce related positions are highly technical as well. Further, our sales personnel highly trained and are important to retaining and growing our businesses. Our success depends in large part upon our ability to identify, hire, retain and motivate highly skilled professionals.
We face intense competition for these professionals from our competitors, customers, marketing partners and other companies throughout the industries in which we compete. Since our inception we have successfully recruited and hired qualified key employees. Any failure on our part to hire, train, and retain a sufficient number of qualified professionals would seriously damage our business.
We depend heavily on the services of our senior management. We believe that our future success depends on the continued services of such management. Our business may be harmed by the loss of a significant number of our senior management in a short period of time.
The insurance we purchase to cover our potential business risk may be inadequate.
Although we believe that our present insurance coverage is sufficient to cover our current estimated exposures, we cannot assure that we will not incur losses or liabilities in excess of our policy limits. In addition, although we believe that will be able to continue to obtain adequate coverage, we cannot assure that we will be able to do so at acceptable costs.
Risks relating to our international operations
Foreign currency exchange rate fluctuations may adversely affect our business.
Since we operate as a multinational corporation that sells and sources products in many different countries, changes in exchange rates could in the future, adversely affect our cash flows and results of operations.
Furthermore, reported sales and purchases made in non-U.S. currencies by our international businesses, when translated into U.S. dollars for financial reporting purposes, fluctuate due to exchange rate movement. Due to the number of currencies involved, the variability of currency exposures and the potential volatility of currency exchange rates, we cannot predict the effect of exchange rate fluctuations on future sales and operating results.
We are subject to economic, political and other risks associated with our significant international business, which could adversely affect our financial results.
We operate internationally primarily through wholly-owned subsidiaries located in North America and Europe. Revenues outside the United States were approximately 9% of total revenues in fiscal 2018. Our sales and earnings could be adversely affected by a variety of factors resulting from our international operations, including
| • | future fluctuations in foreign currency exchange rates; |
| • | complex regulatory requirements and changes in those requirements; |
| • | trade protection measures and import or export licensing requirements; |
| • | multiple jurisdictions and differing tax laws, as well as changes in those laws; |
| • | restrictions on our ability to repatriate investments and earnings from foreign operations; |
| • | changes in the political or economic conditions in a country or region, particularly in developing or emerging markets; |
| • | changes in shipping costs; and |
| • | difficulties in collecting on accounts receivable. |
| 35 |
If any of these risks materialize, we could face substantial increases in costs, the reduction of profit and the inability to do business.
As we expand our commercialization activities outside of the United States, we will be subject to an increased risk of inadvertently conducting activities in a manner that violates the U.S. Foreign Corrupt Practices Act and similar laws. If that occurs, we may be subject to civil or criminal penalties which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We are subject to the U.S. Foreign Corrupt Practices Act (“FCPA”), which prohibits corporations and individuals from paying, offering to pay, or authorizing the payment of anything of value to any foreign government official, government staff member, political party, or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. We are also subject to the UK Anti-Bribery Act, which prohibits both domestic and international bribery, as well as bribery across both public and private sectors.
In the course of establishing and expanding our commercial operations and seeking regulatory approvals outside of the United States, we will need to establish and expand business relationships with various third parties and we will interact more frequently with foreign officials, including regulatory authorities. Expanded programs to maintain compliance with such laws will be costly and may not be effective. Any interactions with any such parties or individuals where compensation is provided that are found to be in violation of such laws could result in substantial fines and penalties and could materially harm our business. Furthermore, any finding of a violation under one country’s laws may increase the likelihood that we will be prosecuted and be found to have violated another country’s laws. If our business practices outside the United States are found to be in violation of the FCPA, UK Anti-Bribery Act or other similar law, we may be subject to significant civil and criminal penalties which could have a material adverse effect on our financial condition and results of operations.
Risks Relating to our Common Stock
Our stock price has been volatile, which could result in substantial losses for investors.
Our common stock is quoted on the New York Stock Exchange, and there has been historical volatility in the market price of our common stock. The trading price of our common stock has been, and is likely to continue to be, subject to significant fluctuations due to a variety of factors, including:
| • | fluctuations in our quarterly operating and earnings per share results; |
| • | the gain or loss of significant contracts; |
| • | the carrying value of our goodwill and intangible assets; |
| • | loss of key personnel; |
| • | announcements of technological innovations or new products by us or our competitors; |
| • | delays in the development and introduction of new products; |
| • | legislative or regulatory changes; |
| • | general trends in the industries we operate; |
| • | recommendations and/or changes in estimates by equity and market research analysts; |
| • | biological or medical discoveries; |
| • | disputes and/or developments concerning intellectual property, including patents and litigation matters; |
| • | public concern as to the safety of new technologies; |
| • | sales of common stock of existing holders; |
| • | securities class action or other litigation; |
| 36 |
| • | developments in our relationships with current or future customers and suppliers and; |
| • | general economic conditions, both in the United States and worldwide. |
In addition, the stock market in general has experienced extreme price and volume fluctuations that have affected the market price of our common stock, as well as the stock of many companies in our industries. Often, price fluctuations are unrelated to operating performance of the specific companies whose stock is affected.
In the past, following periods of volatility in the market price of a company’s stock, securities class action litigation has occurred against the issuing company. If we were subject to this type of litigation in the future, we could incur substantial costs and a diversion of our management’s attention and resources, each of which could have a material adverse effect on our revenue and earnings. Any adverse determination in this type of litigation could also subject us to significant liabilities.
Because we do not intend to pay cash dividends on our common stock, an investor in our common stock will benefit only if it appreciates in value.
We currently intend to retain our retained earnings and future earnings, if any, to finance the expansion of our business and do not expect to pay any cash dividends on our common stock in the foreseeable future. As a result, the success of an investment in our common stock will depend entirely upon any future appreciation. There is no guarantee that our common stock will appreciate in value or even maintain the price at which investors purchased their shares.
It may be difficult for a third party to acquire us, which could inhibit stockholders from realizing a premium on their stock price.
We are subject to the New York anti-takeover laws regulating corporate takeovers. These anti-takeover laws prohibit certain business combinations between a New York corporation and any “interested shareholder” (generally, the beneficial owner of 20% or more of the corporation’s voting shares) for five years following the time that the shareholder became an interested shareholder, unless the corporation’s board of directors approved the transaction prior to the interested shareholder becoming interested.
Our certificate of incorporation, as amended, and by-laws contain provisions that could have the effect of delaying, deferring or preventing a change in control of us that stockholders may consider favorable or beneficial. These provisions could discourage proxy contests and make it more difficult for stockholders to elect directors and take other corporate actions. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock. These provisions include:
| • | a staggered board of directors, so that it would take three successive annual meetings to replace all directors; and |
| • | advance notice requirements for the submission by stockholders of nominations for election to the board of directors and for proposing matters that can be acted upon by stockholders at a meeting. |
Future sales of shares of our common stock or the issuance of securities senior to our common stock could adversely affect the trading price of our common stock and our ability to raise funds in new equity offerings.
We are not restricted from issuing additional common stock, preferred stock or securities convertible into or exchangeable for common stock. Future sales of a substantial number of our shares of common stock or equity-related securities in the public market or privately, or the perception that such sales could occur, could adversely affect prevailing trading prices of our common stock, and could impair our ability to raise capital through future offerings of equity or equity-related securities. No prediction can be made as to the effect, if any, that future sales of shares of common stock or the availability of shares of common stock for future sale will have on the trading price of our common stock.
Our failure to establish and maintain effective internal controls over financial reporting and information technology access could result in material misstatements in our consolidated financial statements, our failure to meet our reporting obligations and cause investors to lose confidence in our reported financial information, which in turn could cause the trading price of our common stock to decline.
Under Section 404 of the Sarbanes-Oxley Act of 2002 and rules promulgated by the SEC, companies are required to conduct a comprehensive evaluation of their internal control over financial reporting. As part of this process, we are required to document and test our internal control over financial reporting; management is required to assess and issue a report concerning our internal control over financial reporting; and our independent registered public accounting firm is required to attest to the effectiveness of our internal control over financial reporting. Our internal control over financial reporting may not prevent or detect misstatements because of its inherent limitations, including the possibility of human error, the circumvention or overriding of controls, or fraud.
| 37 |
Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be prevented or detected timely. Even effective internal controls over financial reporting can provide only reasonable assurance with respect to the preparation and fair presentation of financial statements.
During the financial close for fiscal year 2018 we identified material weaknesses in our internal controls over financial reporting related to (1) insufficient controls to fully and timely take into account changes in the business environment and experience with ultimate collection from third-party payers in the determination of sales allowance amounts, and (2) information technology access controls. A more complete description of this material weakness is included in Item 9A, “Controls and Procedures” in this Form 10-K.
The existence of a material weakness could result in errors in our financial statements that could result in a restatement of financial statements, which could cause us to fail to meet our reporting obligations, lead to a loss of investor confidence and have a negative impact on the trading price of our common stock.
Item 1B. Unresolved Staff Comments
None
The following are the principal facilities of the Company:
| Leased / | Square | |||||||
| Location | Primary use | Segments | owned | footage | ||||
| Farmingdale, NY (Note 1) |
Clinical laboratory and research | Clinical Laboratory Services | Leased | 43,000 | ||||
| Farmingdale, NY | Manufacturing, research, sales and administrative office | Life Sciences, Therapeutics | Owned | 22,000 | ||||
| New York, NY (Note 2) |
Corporate headquarters | Other | Leased | 11,300 | ||||
| Lausen, Switzerland (Note 3) |
Operational headquarters in Europe, including sales and distribution | Life Sciences Products | Leased | 9,626 | ||||
| Ann Arbor, Michigan (Note 4) |
Manufacturing, research, and distribution | Life Sciences Products | Leased | 26,820 |
Note 1 - On October 9, 2015, this lease was amended and extended through March 31, 2027.
Note 2 - In June 2017, the lease, which includes 4,100 square feet under a sublease rental agreement through December 31, 2018, was extended through June 2028.
Note 3 - In October 2017, the lease was amended and extended through August 2018 and was automatically extended for one year.
Note 4 - In March 2009, the lease was amended and extended through May 2021.
We believe the current facilities are suitable and adequate for the Company’s current operating needs for its clinical laboratories services, life science products and therapeutics segments and that the production capacity in various locations is sufficient to manage services and product requirements.
In August 2018, we entered into an agreement to purchase a commercial facility with nearly 36,000 square feet in Farmingdale, NY at a price of approximately $6.0 million. The purchase of this facility extends Enzo’s New York campus to nearly 101,000 square feet, complementing our existing sites in Michigan and Switzerland.
| 38 |
As of July 31, 2018, there are seven cases that are either pending or on appeal, which were originally brought by the Company in the United States District Court for the District of Delaware (“the Court”), alleging patent infringement against various companies. On June 28, 2017, the Court issued an opinion in the Gen-Probe case, granting Gen-Probe’s motion for summary judgment that the asserted claims of the ’180 patent are invalid for nonenablement. The Court entered final judgment of invalidity of the asserted claims of the ‘180 patent on July 19, 2017 in the Gen-Probe and Hologic cases. The Court entered partial final judgment of invalidity of the asserted claims of the ‘180 patent and stayed the remainder of the cases in the Becton Dickinson and Roche cases on July 31, 2017 and August 2, 2017, respectively. The Company filed notices of appeal in each of the Gen-Probe, Hologic, Becton Dickinson, and Roche cases, which were docketed by the United States Court of Appeals for the Federal Circuit (“Federal Circuit”). In the Abbott case, the parties agreed that the Court’s summary judgment ruling in the Gen-Probe case invalidated all of the ’180 patent claims asserted against the Abbott Defendants. On August 15, 2017, the Court granted Abbott’s motion for summary judgment that the asserted claims of the ’405 patent are invalid for nonenablement. On September 1, 2017, the Court entered final judgment of invalidity of the asserted claims of the ‘180 and ‘405 patents for nonenablement in the Abbott case. Enzo subsequently filed a notice of appeal in the Abbott case on September 14, 2017. The Federal Circuit docketed the appeal on September 15, 2017. The Federal Circuit consolidated the appeals from the Abbott, Becton Dickinson, Gen-Probe, Hologic, and Roche litigations (“Consolidated Appeals”). We disagree with the Court’s invalidity decisions regarding the ‘180 and ‘405 patents in the pending cases as set forth in our opening brief in the Consolidated Appeals pending in the Federal Circuit filed on November 28, 2017. In the Consolidated Appeals, we have asked the Federal Circuit to reverse the Court’s grants of final and summary judgment of invalidity of the asserted claims of the ‘180 and ‘405 patents and to remand the cases against Abbott, Becton Dickinson, Gen-Probe, Hologic, and Roche to the Court. Briefing is now complete in the Consolidated Appeals. The parties await the Federal Circuit’s scheduling of an oral argument date for the Consolidated Appeals. In the other two cases involving Hologic, one of the cases is stayed (Hologic II), while the other case (Hologic III) that involves the ‘581 patent is proceeding under the Court’s scheduling order. In Hologic III, the Court granted Enzo’s motion to amend its complaint to add two new defendants, Grifols Diagnostic Solutions, Inc. and Grifols, S.A, to that case. The parties have completed claim construction briefing and a claim construction hearing, but the Court has not issued a claim construction order. The Court amended the scheduling order such that fact discovery will close on October 31, 2019, dispositive motions will be heard on May 7, 2019, and trial will begin on November 18, 2019. Regarding Hologic’s petition requesting institution of an inter partes review proceeding of U.S. Patent No. 6,221,581 (“the ‘581 patent”) filed with the United States Patent and Trademark Office (“PTO”), the Patent Trial and Appeals Board (“the Board”) denied institution of Hologic’s petition on April 18, 2018. On May 18, 2018, Hologic filed with the Board, a request for rehearing of the order denying institution of inter partes review of the ‘581 patent. Enzo filed a brief in response to Hologic’s request for rehearing.
As of July 31, 2018, the Company and Enzo Life Sciences are engaged in litigation in the United States District Court for the Southern District of New York against Roche Diagnostic GmbH and its related company Roche Molecular Systems, Inc. (“Roche”), as declaratory judgment defendants. This case was commenced in May 2004. Roche seeks a declaratory judgment of non-breach of contract and patent invalidity against the Company and Enzo Life Sciences. Roche has also asserted tort claims against the Company and Enzo Life Sciences. The Company and Enzo Life Sciences have asserted breach of contract and patent infringement causes of action against Roche. There has been extensive discovery. In 2011, Roche moved for summary judgment of non-infringement regarding the Company’s patent claims. In 2012, the motion was granted in part and denied in part. In December 2012, Roche moved for summary judgment on the Company’s non-patent claims. Additional discovery was taken and the Company responded to the motions in May 2013. In December 2013, the Court granted in part and denied in part Roche’s summary judgment motion. In October 2014, the Court ordered that damages discovery concerning the Company’s remaining contract and patent claims and Roche’s claims should be completed by the end of January 2015, and expert discovery should be completed following the Court’s claim construction ruling concerning the Company’s patent infringement claim against Roche. Roche dropped its tort claims during damages discovery. On October 2, 2017, the Court issued its claim construction ruling. On September 8, 2018, the Court issued an order (i) directing that motions for summary judgment should be filed on October 10, 2018 and a proposed pretrial order by February 22, 2019, and (ii) scheduling an April 8, 2019 trial. The Company and Enzo Life Sciences intend to vigorously press their remaining claims and contest the claims against them.
The following legal settlements are included in the statement of operations under Legal settlements, net within the Life Science segment for the 2016 period:
| 39 |
The Company and the U.S. Department of Justice reached a settlement agreement to resolve an investigation focused primarily on an alleged failure to collect diagnosis codes from physicians who ordered tests through Enzo Clinical Labs, and recorded a charge of $2.0 million during fiscal year 2014. The settlement amount is being paid with interest over a five-year period. During fiscal year 2016, the Company accrued an additional $1.5 million, in the statement of operations under legal settlements, net within the Clinical Labs segment, due to the Company’s achievement of certain financial milestones. As of July 31, 2018, the total liability for this settlement is $0.4 million and is included in other current liabilities.
In June 2014, the Company, as plaintiff finalized and executed a settlement agreement with PerkinElmer, Inc., and PerkinElmer Health Sciences, Inc. (together, “PerkinElmer”). PerkinElmer paid $7.0 million in escrow pursuant to the agreement because of a former attorney’s charging lien for fees allegedly owed for past services rendered to the Company. In December 2015, the Company entered into a Settlement Agreement with the former attorney pursuant to which the Company and the former attorney resolved their respective claims against each other. In January 2016, the Company received a total of approximately $7.0 million from the escrow referred to above in accordance with the terms of the Settlement Agreement. In October 2015, the Company reached and finalized a settlement with Affymetrix, Inc. in the amount of $6.8 million, net in a patent infringement action brought by the Company. In January 2016, the Company reached and finalized a settlement agreement with Agilent Technologies, Inc. in the amount of $6.1 million, net in a patent infringement action brought by the Company. In May 2016, the Company reached and finalized a settlement with Life Technologies Corporation in the amount of $24.3 million, net in an infringement action brought by the Company. In July 2016, the Company reached and finalized a settlement with Illumina, Inc., in the amount of $14.5 million, net in an infringement action brought by the Company.
There can be no assurance that the Company will be successful in these litigations. Even if the Company is not successful, management does not believe that there will be a significant adverse monetary impact on the Company.
The Company is party to other claims, legal actions, complaints, and contractual disputes that arise in the ordinary course of business. The Company believes that any liability that may ultimately result from the resolution of these matters will not, individually or in the aggregate, have a material adverse effect on its financial position or results of operations
Item 4. Mine Safety Disclosures
Not Applicable
| 40 |
Part II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
The common stock of the Company is traded on the New York Stock Exchange (Symbol: ENZ). The following table sets forth the closing high and low price of the Company’s common stock for the periods indicated as reported on the New York Stock Exchange.
| 2018 Fiscal Year (August 1, 2017 to July 31, 2018): | ||||||||
| High | Low | |||||||
| 1st Quarter | $ | 11.44 | $ | 9.82 | ||||
| 2nd Quarter | $ | 9.99 | $ | 7.36 | ||||
| 3rd Quarter | $ | 7.30 | $ | 5.43 | ||||
| 4th Quarter | $ | 6.78 | $ | 4.26 | ||||
| 2017 Fiscal Year (August 1, 2016 to July 31, 2017): | ||||||||
| High | Low | |||||||
| 1st Quarter | $ | 6.76 | $ | 4.92 | ||||
| 2nd Quarter | $ | 7.48 | $ | 5.81 | ||||
| 3rd Quarter | $ | 8.88 | $ | 6.40 | ||||
| 4th Quarter | $ | 11.79 | $ | 8.75 | ||||
As of September 30, 2018, the Company had approximately 953 stockholders of record of its common stock.
The Company has not paid a cash dividend on its common stock and intends to continue a policy of retaining earnings to finance and build its operations. Accordingly, the Company does not anticipate the payment of cash dividends to holders of common stock in the foreseeable future.
Performance Graph
The graph below compares the five-year cumulative shareholder total return based upon an initial $100 investment (assuming the reinvestment of dividends) for Enzo Biochem, Inc. shares of Common Stock with the comparable return for the New York Stock Exchange Market Value Index and two peer issuer indices selected on an industry basis. The two peer group indices include: (i) 147 biotechnology companies engaged in the research and development of diagnostics substances and (ii) 18 companies engaged in the medical laboratories business. All of the indices include only companies whose common stock has been registered under Section 12 of the Security Exchange Act of 1934 for at least the time frame set forth in the graph.
Management approves the selection of Peer Group companies, adjusting the group based upon our business and changes in the Peer Group companies’ business or the comparability of their metrics. The Peer Group may also be adjusted in the event of mergers, acquisitions, or other significant economic changes.
The total shareholder returns depicted in the graph are not necessarily indicative of future performance. The Performance Graph and related disclosure shall not be incorporated by reference in any filing by the Company under the Securities Act of 1933 of the Securities Act of 1934, except to the extent that the Company specifically incorporates the graph and such disclosure by reference.
| 41 |
COMPARISON OF 5-YEAR CUMULATIVE TOTAL
RETURN AMONG ENZO BIOCHEM, INC.,
NYSE MARKET INDEX, MORNINGSTAR DIAGNOSTIC AND RESEARCH
INDEX AND MEDICAL LABORATORIES INDEX
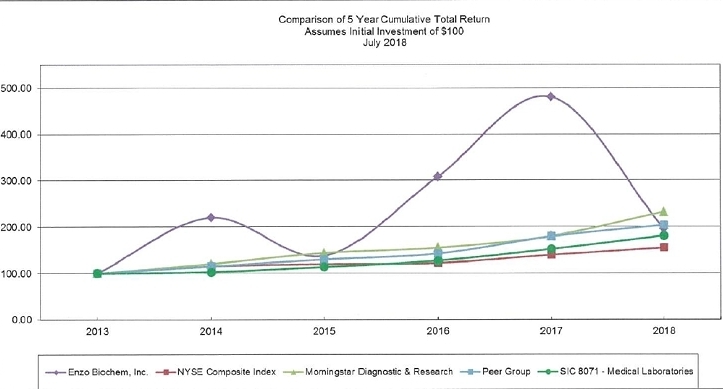
ASSUMES $100 INVESTED ON AUGUST 1, 2013
ASSUMES DIVIDEND REINVESTED
COMPARISON OF CUMULATIVE TOTAL RETURN OF ONE OR MORE
COMPANIES, PEER GROUPS, INDUSTRY INDEXES AND/OR BROAD MARKETS
| 7/31/2013 | 7/31/2014 | 7/31/2015 | 7/31/2016 | 7/31/2017 | 7/31/2018 | |||||||
| Company/Market/Peer Group | ||||||||||||
| Enzo Biochem, Inc. | 100.00 | 220.18 | 137.61 | 308.26 | 479.85 | 195.48 | ||||||
| NYSE Composite Index | 100.00 | 115.09 | 119.67 | 121.99 | 139.00 | 154.57 | ||||||
| Morningstar Diagnostic & Research | 100.00 | 120.33 | 143.79 | 155.03 | 180.07 | 232.29 | ||||||
| Peer Group | 100.00 | 114.97 | 129.70 | 142.34 | 179.05 | 203.49 | ||||||
| Peer Group + Enzo Biochem Inc. | 100.00 | 115.18 | 129.69 | 142.72 | 179.77 | 203.39 | ||||||
| SIC 8071 - Medical Laboratories | 100.00 | 102.84 | 113.67 | 126.97 | 151.99 | 180.58 | ||||||
| Medical Laboratories + Enzo Biochem Inc. | 100.00 | 103.29 | 113.74 | 127.66 | 153.28 | 180.54 |
| 42 |
Item 6. Selected Financial Data
The following table, which is derived from the audited consolidated financial statements of the Company for the fiscal years 2014 through 2018 should be read together with the discussion in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the Company’s consolidated financial statements and notes to those statements included elsewhere in this Annual Report on Form 10-K.
|
For the fiscal year ended
July 31, (In thousands, except per share amounts) |
||||||||||||||||||||
| Operating Results | 2018 | 2017 | 2016 | 2015 | 2014 | |||||||||||||||
| Revenues | $ | 104,713 | $ | 107,804 | $ | 102,773 | $ | 97,599 | $ | 95,947 | ||||||||||
| Operating income (loss) | $ | (12,164 | ) | $ | (3,148 | ) | $ | 46,848 | $ | (1,206 | ) | $ | (10,180 | ) | ||||||
| Net income (loss) (1) | $ | (10,321 | ) | $ | (2,504 | ) | $ | 45,286 | $ | (2,285 | ) | $ | (9,977 | ) | ||||||
| Basic net income (loss) per common share: | $ | (0.22 | ) | (0.05 | ) | 0.98 | (0.05 | ) | (0.23 | ) | ||||||||||
| Diluted net income (loss) per common share: | $ | (0.22 | ) | $ | (0.05 | ) | $ | 0.97 | $ | (0.05 | ) | $ | (0.23 | ) | ||||||
| July 31, | ||||||||||||||||||||
| Financial Position (in thousands) | 2018 | 2017 | 2016 | 2015 | 2014 | |||||||||||||||
| Working capital | $ | 63,014 | $ | 71,274 | $ | 70,829 | $ | 22,528 | $ | 15,771 | ||||||||||
| Total assets | $ | 101,660 | $ | 107,665 | $ | 111,821 | $ | 68,394 | $ | 64,411 | ||||||||||
| Stockholders’ equity | $ | 81,121 | $ | 88,872 | $ | 89,554 | $ | 42,606 | $ | 36,950 | ||||||||||
Notes to Selected Financial Data:
| (1) | In the fiscal year 2016, the Company recorded legal settlements, net of approximately $57.3 million. |
| 43 |
| (2) | Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations |
See in this Form 10-K for the fiscal year ended July 31, 2018 Part 1. Item 1. Business, for Forward Looking Cautionary Statements.
The Company’s Enzo Clinical Laboratory Services and Enzo Life Sciences Products reporting units, as described below, are affected by different US and global economic conditions which are included in Item 1A, Risk Factors.
We are comprised of three operating companies that have evolved out of our core competence: the use of nucleic acids as informational molecules and the use of compounds for immune modulation. These wholly-owned operating companies and the foreign subsidiaries of Enzo Life Sciences conduct their operations through three reportable segments. Below are brief descriptions of each of the three operating segments (see Note 15 in the Notes to Consolidated Financial Statements):
Enzo Clinical Laboratory Services is a regional clinical laboratory serving the greater New York and New Jersey medical communities and expanding into Connecticut. The Company believes having clinical diagnostic services allows us to capitalize first hand on our extensive advanced molecular and cytogenetic capabilities and the broader trends in predictive and personalized diagnostics. We offer a menu of routine and esoteric clinical laboratory tests or procedures used in general patient care by physicians to establish or support a diagnosis, monitor treatment or medication, or search for an otherwise undiagnosed condition. We operate a full-service clinical laboratory in Farmingdale, New York, a network of 32 patient service centers throughout greater New York and New Jersey, a free-standing “STAT” or rapid response laboratory in New York City and Connecticut, and a full-service phlebotomy and an in-house logistics department. Payments for clinical laboratory testing services are made by the Medicare program, healthcare insurers and patients.
The Clinical Laboratory Services reporting unit is impacted by various risk factors, including among others, reduced reimbursements from third party payers for testing performed and from recent health care legislation. Despite the growth we have experienced in previous years, there can be no assurance future growth can be achieved. The introduction of new molecular and esoteric tests is expected to increase our revenue per test and could offset impacts from the above factors. The Company anticipates improved profitability with increased service volume. Clinical Laboratory Services experienced year over year growth in fiscal 2017 and 2016 of 9% and 12%, respectively.
Enzo Life Sciences Products manufactures, develops and markets products and tools to life sciences, drug development and clinical research customers world-wide and has amassed a large patent and technology portfolio. Enzo Life Sciences, Inc. is a recognized leader in labelling and detection technologies across research and diagnostic markets. Our strong portfolio of proteins, antibodies, peptides, small molecules, labelling probes, dyes and kits provides life science researchers tools for target identification/validation, high content analysis, gene expression analysis, nucleic acid detection, protein biochemistry and detection, and cellular analysis. We are globally recognized and acknowledged as a leader in manufacturing, in-licensing, and commercialization of over 40,000 products. Our strategic focus is directed to innovative high quality research reagents and kits in the primary key research areas of genomics, immunohistochemistry, immunoassays, cellular analysis, and small molecule chemistry. The segment is an established source for a comprehensive panel of products to scientific experts in the fields of cancer, cardiovascular disease, neurological disorders, diabetes and obesity, endocrine disorders, infectious and autoimmune disease, hepatotoxicity and renal injury.
Enzo Therapeutics is a biopharmaceutical venture that has developed multiple novel approaches in the areas of gastrointestinal, infectious, ophthalmic and metabolic diseases, many of which are derived from the pioneering work of Enzo Life Sciences. The Company has focused its efforts on developing treatment regimens for diseases and conditions in which current treatment options are ineffective, costly, and/or cause unwanted side effects. This focus has generated a clinical and preclinical pipeline, as well as more than 154 patents and patent applications.
The following table summarizes the sources of revenues for the fiscal years ended July 31, 2018, 2017 and 2016 (in $000’s and percentages):
| 2018 | 2017 | 2016 | ||||||||||||||||||||||
| Clinical laboratory services | $ | 74,777 | 71 | % | $ | 77,407 | 72 | % | $ | 70,915 | 69 | % | ||||||||||||
| Product revenues | 29,224 | 28 | 29,192 | 27 | 30,337 | 30 | ||||||||||||||||||
| Royalty and license fee income | 712 | 1 | 1,205 | 1 | 1,521 | 1 | ||||||||||||||||||
| Total | $ | 104,713 | 100 | % | $ | 107,804 | 100 | % | $ | 102,773 | 100 | % | ||||||||||||
| 44 |
Results of Operations
Fiscal year ended July 31, 2018 compared to July 31, 2017
(in 000s)
| Comparative Financial Data for the Fiscal Years Ended July 31, | ||||||||||||||||
| 2018 | 2017 |
Increase (Decrease) |
% Change | |||||||||||||
| Revenues: | ||||||||||||||||
| Clinical laboratory services | $ | 74,777 | $ | 77,407 | $ | (2,630) | (3 | ) | ||||||||
| Product revenues | 29,224 | 29,192 | 32 | - | ||||||||||||
| Royalty and license fee income | 712 | 1,205 | (493 | ) | (41 | ) | ||||||||||
| Total revenues | 104,713 | 107,804 | (3,091 | ) | (3 | ) | ||||||||||
| Operating costs and expenses: | ||||||||||||||||
| Cost of clinical laboratory services | 46,008 | 45,400 | 608 | 1 | ||||||||||||
| Cost of product revenues | 14,377 | 14,078 | 299 | 2 | ||||||||||||
| Research and development | 3,210 | 2,928 | 282 | 10 | ||||||||||||
| Selling, general and administrative | 44,465 | 44,092 | 373 | 1 | ||||||||||||
| Provision for uncollectible accounts receivable | 3,690 | 2,775 | 915 | 33 | ||||||||||||
| Legal fee expense | 5,127 | 1,679 | 3,448 | ** | ||||||||||||
| Total operating costs and expenses | 116,877 | 110,952 | 5,925 | 5 | ||||||||||||
| Operating loss | (12,164 | ) | (3,148 | ) | (9,016 | ) | ** | |||||||||
| Other income (expense): | ||||||||||||||||
| Interest | 853 | 384 | 469 | ** | ||||||||||||
| Other | 168 | 125 | 43 | 34 | ||||||||||||
| Foreign currency (loss) gain | (275 | ) | 135 | (410 | ) | ** | ||||||||||
| Loss before income taxes | $ | (11,418 | ) | $ | (2,504 | ) | $ | 8,914 | ** | |||||||
** not meaningful
Consolidated Results:
The “2018 period” and the “2017 period” refer to the fiscal year ended July 31, 2018 and 2017, respectively.
Clinical laboratory services revenues for the 2018 period were $74.8 million compared to $77.4 million in the 2017 period, a decrease of $2.6 million or 3% due to lower insurance payments and shifts in test mix to lower esoteric testing versus high genetic testing in the prior year. Total diagnostic testing volume, measured by the number of accessions reported, increased 4% year over year.
Product revenues for the 2018 and 2017 periods was $29.2 million. During the 2018 period, we experienced lower product order volume from customers of $0.5 million, primarily due to competition in the United States, which was offset by the positive impact of foreign currency translation of $0.5 million.
The cost of clinical laboratory services during the 2018 period was $46.0 million as compared to $45.4 million in the 2017 period, an increase of $0.6 million or 1%. The increase is comprised of $1.2 million for reagents, $1.4 million in compensation related expenses, offset by a decrease of $2.1 million for outside reference lab testing costs due to lower genetic testing ordering and internalizing the use of our AMPIPROBE® technology platform. Gross profit margin was 39% in the 2018 period and 41% in the 2017 period, impacted by the mix of tests and decreased payer reimbursement rates.
The cost of product revenues was $14.4 million in the 2018 period and $14.1 million in the 2017 period, an increase of $0.3 million or 2% due to the sale of lower margin items. The gross profit margin on products was 51% in the 2018 period and 52% in the 2017 period.
Research and development expenses were $3.2 million in the 2018 period versus $2.9 million in the 2017 period, an increase of $0.3 million or 10%. The expense for the Life Sciences segment was $2.3 million in both periods. The expense for the Therapeutics segment was $0.9 million in the 2018 period and $0.6 million in the 2017 period. The lower expense in the 2017 period was due to the impact of an adjustment to a previously recorded obligation for clinical trial activity no longer necessary.
| 45 |
Selling, general and administrative expenses were approximately $44.5 million during the 2018 period versus $44.1 million during the 2017 period, an increase of $0.4 million or 1%. The Clinical Laboratory services segment expense increased $0.2 million, primarily for customer services related salary expenses of $0.8 million, partially offset by a reduction of $0.6 million in sales salaries and commissions. The Life Sciences segment expense increased $0.4 million due to the impact of an adjustment recorded in the 2017 period which decreased a royalty related obligation that was no longer required. The Other segment expense decreased $0.2 million, primarily due to a decrease in compensation related expenses.
The provision for uncollectible accounts receivable, primarily related to the Clinical Labs segment, was approximately $3.7 million in the 2018 period and $2.8 million in the 2017 period, an increase of $0.9 million or 33%. As a percentage of Clinical laboratory services, the provision for uncollectible accounts receivable relating to the Clinical Labs segment was 4.9% in the 2018 period and 3.5% in the 2017 period.
Legal fee expense was $5.1 million during the 2018 period compared to $1.7 million in the 2017 period, an increase of $3.4 million due to the timing of legal activity and related costs associated with on-going litigation and contract dispute where the Company is the plaintiff.
Segment Results:
Clinical Laboratory Services
Revenue from laboratory services for the 2018 period were $74.8 million compared to $77.4 million in the 2017 period. The decrease of $2.6 million or 3% is attributed to the loss of a large medical practice that internalized its genetic testing ordering, storm related weather in the Northeast, and by decreased payer reimbursement rates. Cost of services during the 2018 period was $46.0 million as compared to $45.4 million in the 2017 period, an increase of $0.6 million or 1%. Gross profit margin was 39% in the 2018 period and 41% in the 2017 period and was impacted by the mix of tests and by decreased payer reimbursement rates. As a percentage of revenues, the provision for uncollectable accounts, primarily for self-pay patient accounts, was 4.9% for the 2018 period and 3.5% for the 2017 period. Income before taxes was $0.3 million for 2018 period as compared to $4.7 million in the 2017 period, a decrease of $4.4 million.
Life Sciences Products
Product revenues for the 2018 and 2017 periods were $29.2 million. During the 2018 period, we experienced lower product order volume of $0.5 million, primarily due to lower research funding and lower pricing due to competition in the United States, which was offset by the positive impact of foreign currency translation of $0.5 million. The segment’s gross profit was $15.6 million in the 2018 period and $16.3 million in the 2017 period, primarily due to the decline in royalty income. The license agreement on which royalties were earned expired in the 2018 period. The gross profit margin on products was 51% in the 2018 period and 52% in the 2017 period and was negatively impacted by the sale of lower margin products. In the 2018 period, selling general and administrative expenses increased $0.4 million compared to the 2017 period. Due to the impacts of Swiss franc depreciation versus the US dollar and Euro and British pound sterling depreciation versus the Swiss franc during the 2018 period, the foreign currency loss was $0.3 million compared to a gain of $0.1 million in the 2017 period, an unfavorable change of $0.4 million. Income before taxes was $1.4 million for the 2018 period versus $2.8 million for the 2017 period.
Therapeutics
The Therapeutics segment’s operating loss before income taxes was approximately $0.9 million in the 2018 period and $0.6 million in the 2017 period. The lower loss during the 2017 period was due to the impact of an adjustment to a previously recorded obligation for clinical trial activity no longer necessary.
Other
The Other segment’s loss before taxes for the 2018 period was approximately $12.2 million compared to $9.3 million for the 2017 period, an increase of $2.9 million. During the 2018 period, legal fee expense associated with on-going litigation and contract dispute increased $3.5 million. The 2018 period selling general and administrative expense declined $0.2 million compared to the 2017 period primarily due to a decrease in compensation related expenses. Interest income increased $0.4 million in the 2018 period due to the impact of rising interest rates earned on cash and cash equivalents and because of interest expense incurred in the 2017 period on a loan which was repaid during that period.
| 46 |
Results of Operations
Fiscal year ended
July 31, 2017 compared to July 31, 2016
(in 000s)
| Comparative Financial Data for the Fiscal Years Ended July 31, | ||||||||||||||||
| 2017 | 2016 |
Increase (Decrease) |
% Change | |||||||||||||
| Revenues: | ||||||||||||||||
| Clinical laboratory services | $ | 77,407 | $ | 70,915 | $ | 6,492 | 9 | |||||||||
| Product revenues | 29,192 | 30,337 | (1,145 | ) | (4 | ) | ||||||||||
| Royalty and license fee income | 1,205 | 1,521 | (316 | ) | (21 | ) | ||||||||||
| Total revenues | 107,804 | 102,773 | 5,031 | 5 | ||||||||||||
| Operating costs, expenses and legal settlements, net: | ||||||||||||||||
| Cost of clinical laboratory services | 45,400 | 42,859 | 2,541 | 6 | ||||||||||||
| Cost of product revenues | 14,078 | 14,331 | (253 | ) | (2 | ) | ||||||||||
| Research and development | 2,928 | 3,524 | (596 | ) | (17 | ) | ||||||||||
| Selling, general, and administrative | 44,010 | 43,586 | 424 | 1 | ||||||||||||
| Provision for uncollectible accounts receivable | 2,775 | 2,336 | 439 | 19 | ||||||||||||
| Legal fee expense | 1,679 | 6,384 | (4,705 | ) | (74 | ) | ||||||||||
| Legal settlements, net | — | (57,250 | ) | 57,250 | ** | |||||||||||
| Total costs, expenses and legal settlements, net | 110,870 | 55,770 | 55,100 | 99 | ||||||||||||
| Operating income (loss) | (3,066 | ) | 47,003 | (50,069 | ) | ** | ||||||||||
| Other income (expense): | ||||||||||||||||
| Interest | 384 | (136 | ) | 520 | ** | |||||||||||
| Other | 125 | 122 | 3 | 2 | ||||||||||||
| Foreign currency loss | 135 | (474) | 609 | ** | ||||||||||||
| Income (loss) before income taxes | $ | (2,422 | ) | $ | 46,515 | $ | (48,937 | ) | ** | |||||||
** not meaningful
Consolidated Results:
The “2017 period” and the “2016 period” refer to the fiscal year ended July 31, 2017 and 2016, respectively.
Clinical laboratory services revenues for the 2017 period were $77.4 million compared to $70.9 million in the 2016 period, an increase of $6.5 million or 9%. The increase is attributed to molecular testing volume in women’s health markets, the addition of new accounts, and expansion of the service area versus the 2016 period.
Product revenues for the 2017 period were $29.2 million compared to $30.3 million in the 2016 period, a decrease of $1.1 million or 4%. The decrease resulted from lower product order volume due to lower research funding and from lower pricing due to competition in both the United States and in foreign markets totaling $0.8 million, and the negative impact of foreign currency translation of $0.3 million.
The cost of clinical laboratory services during the 2017 period was $45.4 million as compared to $42.9 million in the 2016 period, an increase of $2.5 million or 6% due to the volume increase in clinical laboratory services revenue from molecular testing.
The cost of product revenues was $14.1 million in the 2017 period and $14.3 million in the 2016 period, a decrease of $0.2 million or 1% due to lower product revenues. The gross profit margin was 52% in the 2017 period and 53% in the 2016 period.
Research and development expenses were $2.9 million versus $3.5 million in the 2016 period, a decrease of $0.6 million or 17%. The expense for the Life Sciences segment decreased $0.4 million due to lower compensation, materials and patent expenses. The expense for the Therapeutics segment decreased $0.2 million due to the impact of an adjustment decreasing an obligation for clinical trial activity.
| 47 |
Selling, general and administrative expenses were approximately $44.0 million during the 2017 period versus $43.6 million during the 2016 period, an increase of $0.4 million or 1%. The Clinical Lab segment expense increased $1.6 million comprised of sales and support compensation costs of $0.6 million, miscellaneous office, information technology and business development expenses of $0.8 million, and collection expenses for self-pay patient receivables of $0.2 million. The Life Sciences segment expense decreased $0.5 million due to a decrease of $0.4 million from the adjustment of an obligation relating to a lawsuit in which we were the plaintiff and $0.1 million in salaries. The Other segment expense decreased $0.6 million. During the 2016 period, we incurred consulting and printing expenses related to the contested proxy for the 2016 annual stockholders’ meeting of $1.2 million, partially offset by higher compensation expenses of $0.5 million and professional fees of $0.1 million in the 2017 period.
The provision for uncollectible accounts receivable, primarily related to the Clinical Labs segment, was approximately $2.8 million in the 2017 period and $2.3 million in the 2016 period, an increase of approximately $0.4 million. As a percentage of Clinical laboratory services, the provision for uncollectible accounts receivable relating to the Clinical Labs segment was 3.5% in the 2017 period and 3.3% in the 2016 period.
Legal fee expense was $1.7 million during the 2017 period compared to $6.4 million in the 2016 period, a decrease of $4.7 million or 74% due to the timing of legal activity and related costs associated with on-going patent litigation where the Company is plaintiff. Legal fee expense in the 2016 period also included $0.4 million for contested proxy costs relating to our 2016 annual stockholders’ meeting.
There were no legal settlements during the 2017 period. Legal settlements, net was $57.3 million in the 2016 period. During the 2016 period the Company as plaintiff finalized and executed settlement agreements with Affymetrix, Inc., Agilent Technologies, Inc., Life Technologies Corporation, and Illumina Inc. relating to patent infringement claims and collected proceeds held in escrow relating to the PerkinElmer, Inc. and Molecular Probes, Inc. settlements, all totalling $58.8 million of net proceeds. The Company also recorded an additional charge of $1.5 million relating to the 2014 settlement with the U.S. Department of Justice, due to the achievement of certain financial milestones.
Segment Results:
Clinical Laboratory Services
Revenue from laboratory services for the 2017 period were $77.4 million compared to $70.9 million in the 2016 period. The increase of $6.5 million or 9% is attributed to increased molecular testing volume and the addition of new accounts and expansion of the service area. Cost of sales during the 2017 period was $45.4 million as compared to $42.9 million in the 2016 period, an increase of $2.5 million or 6% due to higher testing volume. Gross profit margin was 41% in the 2017 period and 40% in the 2016 period attributed to higher margin molecular testing. As a percentage of revenues, the provision for uncollectable accounts was 3.5% for the 2017 period and 3.3% for the 2016 period. Income before taxes was $4.7 million for 2017 period as compared to $1.1 million in the 2016 period, an increase of $3.6 million. The 2016 period includes an additional $1.5 million charge for the legal settlement with the U.S. Department of Justice, due to the achievement of certain financial milestones.
Life Sciences Products
Product revenues for the 2017 period were $29.1 million compared to $30.3 million in the 2016 period, a decrease of $1.1 million or 4%. The decrease is due to lower product sales of $0.8 million in the United States and foreign markets and the negative impact of foreign currency translation of $0.3 million. The segment’s gross profit was $16.3 million in the 2017 period and $17.5 million in the 2016 period. Due to appreciation of foreign currencies versus the US dollar, in particular the Euro during the 2017 period versus the 2016 period, the foreign currency gain was $0.1 million compared to a loss of $0.5 million in the 2016 period, a favorable change of $0.6 million. Income before taxes was $2.8 million for the 2017 period as compared to $61.4 million for the 2016 period, a decrease of $58.6 million. The 2016 period includes $58.8 million for patent litigation settlements previously described.
Therapeutics
The Therapeutics segment’s operating loss before income taxes was approximately $0.6 million and $0.8 million in the 2017 and 2016 periods, respectively, a decrease of $0.2 million due to the impact of an adjustment decreasing an obligation for clinical trial activity.
Other
The Other segment’s operating loss before taxes for the 2017 period was approximately $9.3 million as compared to $15.2 million for the 2016 period, an improvement of $5.9 million. During the 2017 period, legal fee expense associated with on-going patent litigation declined $4.4 million. Interest income increased $0.5 million due to the impacts of both the higher level of cash and cash equivalents earning interest in the 2017 period and the repayment of the loan payable during the beginning of the second quarter of 2017 period.
| 48 |
The 2016 period included $1.6 million of consulting, legal fee and printing expenses relating to contested proxy costs for the 2016 annual stockholders’ meeting. These favorable impacts were partially offset by an increase in compensation and benefits expenses of $0.5 million and professional fees of $0.1 million in the 2017 period.
Liquidity and Capital Resources
At July 31, 2018, the Company had cash and cash equivalents of $60.0 million of which $0.4 million was in foreign accounts, as compared to cash and cash equivalents of $64.2 million, of which $0.5 million was in foreign accounts at July 31, 2017. It is the Company’s current intent to permanently reinvest these funds outside of the United States, and its current plans do not demonstrate a need to repatriate them to fund its United States operations. The Company had working capital of $63.0 million at July 31, 2018 compared to $71.3 million at July 31, 2017. The decrease in working capital of $8.3 million was primarily due to the period loss and net changes in operating assets and liabilities.
Net cash used in operating activities in fiscal 2018 was approximately $2.7 million as compared to net cash used in operating activities of $0.2 million in fiscal 2017, an increase of approximately $2.5 million. The increase in net cash used in operating activities in the 2018 period versus the 2017 period was primarily due to an increase in net loss of $7.8 million, offset by a net change in adjustments and operating assets and liabilities of $5.3 million. Net cash used in operating activities in fiscal 2017 was approximately $0.2 million as compared to net cash provided by operating activities of $53.1 million in fiscal 2016, a decrease of approximately $53.3 million. The decrease in net cash provided by operating activities in the 2017 period versus the 2016 period was primarily due to a decrease in net income of $47.8 million, derived principally from the legal settlements reached during that period, and a net change in adjustments and operating assets and liabilities of $5.5 million.
Net cash used in investing activities in fiscal 2018 was approximately $1.9 million as compared to $1.7 million in the year ago period, an increase of $0.2 million. The increase in the 2018 period is primarily due to capital expenditures. Net cash used in investing activities in fiscal 2017 was approximately $1.7 million as compared to $1.5 million in fiscal 2016, an increase of $0.2 million due to capital expenditures.
Net cash provided in financing activities in fiscal 2018 was approximately $0.6 million as compared to cash used of $1.7 million in fiscal 2017. The decrease of $2.3 million was due to the repayment in full of the Credit Agreement in fiscal 2017, and by an increase of $0.4 million in proceeds from the exercise of stock options. Net cash used in financing activities in fiscal 2017 was approximately $1.7 million as compared to $1.9 million in fiscal 2016. The decrease of $0.2 million was due to an increase in net payments under the Credit Agreement which is now expired, offset by an increase of $0.4 million in proceeds from the exercise of stock options.
In June 2013, the Company entered into a secured Revolving Loan and Security Agreement (the “Credit Agreement”) among the Company and certain of its subsidiaries and MidCap Financial Services, LLC (formerly Healthcare Finance Group, LLC). The Credit Agreement expired and was repaid in full on December 7, 2016.
The Company believes that its current cash and cash equivalents level, and utilization of the Controlled Equity Offering program if necessary, as disclosed in Form 10-K Note 10 to the financial statements are sufficient for its foreseeable liquidity and capital resource needs over at least the next twelve (12) months, although there can be no assurance that future events will not alter such view. Although there can be no assurances, in the event additional capital is required, the Company believes it has the ability to raise additional funds through equity offerings or other sources. Our liquidity plans are subject to a number of risks and uncertainties, including those described in the Item 1A. “Risk Factors” section of our Form 10-K for the year ended July 31, 2018, some of which are outside our control. Macroeconomic conditions could limit our ability to successfully execute our business plans and therefore adversely affect our liquidity plans.
Effect of New Accounting Pronouncements
Recently Adopted Accounting Pronouncements
In March 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2016-09, Improvements to Employee Share-Based Payment Accounting, which requires all excess tax benefits or deficiencies to be recognized as income tax expense or benefit in the income statement. In addition, excess tax benefits should be classified along with other income tax cash flows as an operating activity in the statement of cash flows. We adopted this standard in the fiscal year ended July 31, 2018. We recognize compensation expense by amortizing the fair values of awards on a straight line basis over the vesting period, adjusted for forfeitures when they occur. The adoption of this standard did not have a material impact on our consolidated financial statements.
In May 2018, the FASB issued ASU No. 2018-05, Income Taxes (Topic 740): Amendments to SEC Paragraphs Pursuant to SEC Staff Accounting Bulletin No. 118, regarding the accounting implications of the recently issued Tax Cuts and Jobs Act (the “Act”). This standard is effective immediately. The update clarifies that in a company's financial statements that include the reporting period in which the Act was enacted, the company must first reflect the income tax effects of the Act in which the accounting under GAAP is complete. These amounts would not be provisional amounts. The company would also report provisional amounts for those specific
| 49 |
income tax effects for which the accounting under GAAP is incomplete but a reasonable estimate can be determined. We have recorded a provisional amount which we believe is a reasonable estimate of the effects of the Act on our financial statements as of July 31, 2018. Technical corrections or other forthcoming guidance could change how we interpret provisions of the Act, which may impact our effective tax rate and could affect our deferred tax assets, tax positions and/or our tax liabilities.
Pronouncements Issued but Not Yet Adopted
In May 2014, FASB issued ASU No. 2014-09, Revenue from Contracts with Customers: Topic 606. This ASU and its amendments supersede existing revenue recognition guidance, including industry-specific guidance. The core principle of the revenue recognition standard is to require an entity to recognize as revenue the amount that reflects the consideration which it expects to be entitled to in exchange for the goods or services it transfers control of to its customers.
We will adopt this ASU in the first quarter of our fiscal year beginning August 1, 2018 using the full retrospective method. We continue to assess the impact of this ASU on our results of operations, financial position, cash flows and disclosures. Based on our assessment of this ASU, the majority of the amounts that were historically classified as the provision for uncollectible accounts receivable, primarily related to patient responsibility, will be considered an implicit price concession in determining revenues from clinical laboratory services. Accordingly, we will report uncollectible balances associated with patient responsibility as a reduction of the transaction price and therefore as a reduction in revenues from clinical laboratory services, when historically these amounts were classified as the provision for uncollectible accounts receivable within operating costs and expenses. The residual balance of the provision for uncollectible accounts receivable will also be reclassified and included in selling, general and administrative expense. As a result of the adoption of this ASU, we preliminarily estimate the following impact to our consolidated statements of operations for the years ended July 31, 2018 and 2017:
| Year Ended July 31, 2018 | Year Ended July 31, 2017 | ||||||||
| As Reported |
Adjustment for ASU on Revenue Recognition |
Reclassification
of Residual |
As
Adjusted |
As Reported |
Adjustment for ASU on Revenue Recognition |
Reclassification
of Residual |
As
Adjusted |
||
| Total Revenues | $104,713 | $(3,700) | - | $101,013 | $107,804 | $(2,718) | - | $105,086 | |
| Provision for uncollectible accounts receivable | 3,690 | (3,700) | $10 | - | 2,775 | (2,718) | $(57) | - | |
| Selling, general and administrative expenses | 44,435 | - | (10) | 44,425 | 44,092 | - | 57 | 44,149 | |
| Net loss | $(10,321) | - | - | $(10,321) | $(2,504) | - | - | $(2,504) | |
In addition, the adoption of this ASU will result in increased disclosure, including qualitative and quantitative disclosures about the nature, amount, timing and uncertainty of revenue and cash flows arising from contracts with customers. However, the adoption of this ASU is not expected to have a material impact on our financial position or cash flows.
In February 2016, FASB issued ASU No. 2016-02 – Leases (Topic 842). The new standard establishes a right-of-use (ROU) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the income statement. The new standard is effective for our fiscal year beginning August 1, 2019 including interim periods within that fiscal year. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. We believe the adoption of this standard would materially impact our consolidated financial statements by significantly increasing our non-current assets and non-current liabilities on our consolidated balance sheets if we record the right of use assets and related lease
| 50 |
liabilities for our existing operating leases. We will recognize expense in the consolidated statement of operations similar to current lease accounting, in the cost of sales and selling, general and administrative.
In July 2018, FASB issued ASU No. 2018-11 – Leases (Topic 842) – Targeted Improvements. The amendments in this Update provide entities with an additional and optional transition method to adopt the new leases standard. Under this new transition method, an entity initially applies the new leases standard at the adoption date and recognizes a cumulative-effect adjustment to the opening balance of retained earnings in the period of adoption. Consequently, an entity’s reporting for the comparative periods presented in the financial statements in which it adopts the new leases standard will continue to be in accordance with current GAAP (Topic 840, Leases). An entity that elects this additional and optional transition method must provide the required Topic 840 disclosures for all periods that continue to be in accordance with Topic 840. The amendments do not change the existing disclosure requirements in Topic 840. We are currently assessing the impact of the adoption of the amendments in this Update on our results of operations, financial position and cash flows.
In June 2016, FASB issued ASU No. 2016-13 Financial Instruments – Credit Losses (Topic 326). This standard changes the impairment model for most financial instruments, including trade receivables, from an incurred loss method to a new forward-looking approach, based on expected losses. The estimate of expected credit losses will require entities to incorporate considerations of historical information, current information and reasonable and supportable forecasts. Adoption of this standard is required for our annual and interim periods beginning August 1, 2020 and must be adopted using a modified retrospective transition approach. We are currently assessing the impact of the adoption of this standard on our results of operations, financial position and cash flows.
In May 2017, the FASB issued ASU 2017-09, Compensation – Stock Compensation (Topic 708) Scope of Modification Accounting which provides guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. Adoption of this standard is required for our annual and interim periods beginning August 1, 2018 with the amendments in the update applied prospectively to an award modified on or after the adoption date. Based on our preliminary assessment of the standard, we expect that any excess income tax benefits or deficiencies from stock-based compensation, which would be recognized as discrete items within income tax expense rather than additional paid in capital, would be offset by an equivalent adjustment to the deferred tax valuation allowance. Accordingly, we expect that the adoption of this standard will have no impact on our reported operations for the foreseeable future. In addition, we expect to continue to account for award forfeitures in the period they occur.
We reviewed all other recently issued accounting pronouncements and have concluded they are not applicable or not expected to be significant to the accounting for our operations.
Contractual Obligations
The Company has entered into various real estate and equipment operating leases and has employment agreements with certain executive officers. The real estate lease for the Company’s Farmingdale Clinical Lab and Research facility is with a related party. See Item 2, Properties, and Note 13 to the Consolidated Financial Statements for a further description of these various leases.
The following is a summary of future payments under the Company’s contractual obligations as of July 31, 2018:
| Payments Due by Period (In thousands) | Total |
Less than 1 year |
1-3 years | 4-5 years |
More than 5 years |
|||||||||||||||
| Operating lease obligations | $ | 33,626 | $ | 6,809 | $ | 8,645 | $ | 5,877 | $ | 12,295 | ||||||||||
| Current and long term debt obligations | 426 | 424 | 2 | — | — | |||||||||||||||
| Employment agreements | 2,498 | 1,153 | 1,345 | — | — | |||||||||||||||
| Capital lease obligations | 543 | 173 | 370 | — | — | |||||||||||||||
| Total | $ | 37,093 | $ | 8,559 | $ | 10,362 | $ | 5,877 | $ | 12,295 | ||||||||||
Management is not aware of any material claims, disputes or settled matters concerning third-party reimbursements that would have a material effect on our financial statements.
Off-Balance Sheet Arrangements
The Company does not have any “off-balance sheet arrangements” as such term is defined in Item 303(a) (4) of Regulation S-K.
| 51 |
Critical Accounting Policies
General
The Company’s discussion and analysis of its financial condition and results of operations are based upon Enzo Biochem, Inc.’s consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires the Company to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses. These estimates and judgments also affect related disclosure of contingent assets and liabilities.
On an on-going basis, we evaluate our estimates, including those related to contractual expense, allowance for uncollectible accounts, inventory, intangible assets and income taxes. The Company bases its estimates on experience and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Product revenues
Revenues from product sales are recognized when the products are shipped and title transfers, typically upon shipment by common carrier.
Royalties
Royalty revenues are recorded in the period earned. Royalties received in advance of being earned are recorded as deferred revenues.
Revenues - Clinical laboratory services
Revenues from the Clinical Laboratory Services segment are recognized upon completion of the testing process for a specific patient and reported to the ordering physician. These revenues and the associated accounts receivable are based on gross amounts billed or billable for services rendered, net of a contractual adjustment, which is the difference between amounts billed to payers and the expected approved reimbursable settlements from such payers.
The following table represents the Clinical Laboratory Services segment’s net revenues and percentages by revenue category (in thousands):
|
Year ended July 31, 2018 |
Year ended July 31, 2017 |
Year ended July 31, 2016 |
|||||||||||||||||||||||
| Category | Revenue | % | Revenue | % | Revenue | % | |||||||||||||||||||
| Third-party payers | $ | 41,370 | 56 | $ | 43,059 | 56 | $ | 34,454 | 49 | ||||||||||||||||
| Medicare | 12,111 | 16 | 12,705 | 16 | 11,392 | 16 | |||||||||||||||||||
| HMO’s | 11,359 | 15 | 10,263 | 13 | 10,325 | 14 | |||||||||||||||||||
| Patient self-pay | 9,937 | 13 | 11,380 | 15 | 14,744 | 21 | |||||||||||||||||||
| Total | $ | 74,777 | 100 | % | $ | 77,407 | 100 | % | $ | 70,915 | 100 | % | |||||||||||||
The Company provides services to certain patients covered by various third-party payers, including the Federal Medicare program. Laws and regulations governing Medicare are complex and subject to interpretation for which action for noncompliance includes fines, penalties and exclusion from the Medicare programs. See Item 3. Legal Proceedings.
Other than the Medicare program, one provider whose programs are included in the “Third-party payers” and “Health Maintenance Organizations” (“HMO’s”) categories represent approximately 39%, 39% and 30% of the Clinical Laboratory Services segment net revenue for the years ended July 31, 2018, 2017 and 2016 respectively.
The Company currently uses one third party reference lab for certain clinical laboratory services we provide which represents 12% of the consolidated purchases for the year ended July 31, 2018. Although there are a limited number of reference labs available for those clinical laboratory services, we believe that these other reference labs could provide us with such testing results on comparable terms. A change in reference labs, however, could cause a delay in obtaining and reporting those test results and a possible loss of services revenues, which would affect operating results adversely.
| 52 |
Contractual Adjustment
The Company’s estimate of contractual adjustment is based on significant assumptions and judgments, such as its interpretation of payer reimbursement policies, and bears the risk of change. The estimation process is based on the experience of amounts approved as reimbursable and ultimately settled by payers, versus the corresponding gross amount billed to the respective payers. The contractual adjustment is an estimate that reduces gross revenue based on gross billing rates to amounts expected to be approved and reimbursed. Gross billings are based on a standard fee schedule we set for all third party payers, including Medicare, health maintenance organizations (“HMO’s”) and managed care. The Company adjusts the contractual adjustment estimate quarterly, based on its evaluation of current and historical settlement experience with payers, industry reimbursement trends, and other relevant factors. The other relevant factors that affect our contractual adjustment include the monthly and quarterly review of: 1) current gross billings and receivables and reimbursement by payer, 2) current changes in third party arrangements and 3) the growth of in-network provider arrangements and managed care plans specific to our Company.
Our clinical laboratory services business is primarily dependent upon reimbursement from third-party payers, such as Medicare (which principally serves patients 65 and older) and insurers. We are subject to variances in reimbursement rates among different third-party payers, as well as constant changes of reimbursement rates. Changes that decrease reimbursement rates or coverage would negatively impact our revenues. The number of individuals covered under managed care contracts or other similar arrangements has grown over the past several years and may continue to grow in the future. In addition, Medicare and other government healthcare programs continue to shift to managed care. These trends will continue to reduce our revenues.
During the years ended July 31, 2018, 2017 and 2016, the contractual adjustment percentages, determined using current and historical reimbursement statistics, were approximately 85%, 84% and 84%, respectively, of gross billings. The Company believes a decline in reimbursement rates or a shift to managed care, or similar arrangements may be offset by the positive impact of an increase in the number of tests we perform. However, there can be no assurance that we can increase the number of tests we perform or that if we do increase the number of tests we perform, that we can maintain that higher number of tests performed, or that an increase in the number of tests we perform would result in increased revenue.
The Company estimates (by using a sensitivity analysis) that each 1% point change in the contractual adjustment percentage could result in a change in clinical laboratory services revenues of approximately $5.0 million, $4.8 million, and $4.5 million, for the years ended July 31, 2018, 2017, and 2016, respectively, and a change in the net accounts receivable of approximately $0.5 and $0.6 million as of July 31, 2018 and 2017, respectively.
Our clinical laboratory financial billing system records gross billings using a standard fee schedule for all payers and does not record contractual adjustment by payer at the time of billing. Therefore, we are unable to quantify the effect of contractual adjustment recorded during the current period that relate to revenue recorded in a previous period. However, we can reasonably estimate our monthly contractual adjustment to revenue on a timely basis based on our quarterly review process, which includes:
| • | an analysis of industry reimbursement trends; |
| • | an evaluation of third-party reimbursement rates changes and changes in reimbursement arrangements with third-party payers; |
| • | a rolling monthly analysis of current and historical claim settlement and reimbursement experience statistics with payers; |
| • | an analysis of current gross billings and receivables by payer. |
Accounts Receivable and Allowance for Doubtful Accounts
Accounts receivable are reported at realizable value, net of allowances for doubtful accounts, which is estimated and recorded in the period of the related revenue.
The following is a table of the Company’s net accounts receivable by segment. The Clinical Laboratory Services segment’s net receivables are detailed by billing category and as a percent to its total net receivables. As of July 31, 2018 and 2017, approximately 74% and 75% respectively, of the Company’s net accounts receivable relates to its Clinical Laboratory Services business, which operates in the New York, New Jersey and Connecticut medical communities. The Life Sciences products segment’s accounts receivable, of which $1.1 million or 32% and $1.1 million or 29% represents foreign receivables as of July 31, 2018 and 2017, respectively, includes royalty receivables of $0 million and $0.4 million, respectively, from Qiagen Corporation.
| 53 |
Net accounts receivable (in thousands)
| July 31, 2018 | July 31, 2017 | |||||||||||||||
| Clinical Labs (by billing category) | Total Amount | % | Total Amount | % | ||||||||||||
| Third party payers | $ | 4,692 | 48 | $ | 7,256 | 64 | ||||||||||
| Patient self-pay | 2,010 | 20 | 1,591 | 14 | ||||||||||||
| Medicare | 1,740 | 18 | 1,385 | 12 | ||||||||||||
| HMO’s | 1,329 | 14 | 1,169 | 10 | ||||||||||||
| Total Clinical Labs | 9,771 | 100 | % | 11,401 | 100 | % | ||||||||||
| Total Life Sciences | 3,376 | 3,779 | ||||||||||||||
| Total accounts receivable – net | $ | 13,147 | $ | 15,180 | ||||||||||||
Changes in the Company’s allowance for doubtful accounts are as follows:
| July 31, 2018 | July 31, 2017 | |||||||
| Beginning balance | $ | 3,576 | $ | 3,517 | ||||
| Provision for doubtful accounts | 3,690 | 2,775 | ||||||
| Write-offs, net | (4,598 | ) | (2,716 | ) | ||||
| Ending balance | $ | 2,668 | $ | 3,576 | ||||
For the Clinical Laboratory Services segment, the allowance for doubtful accounts represents amounts that the Company does not expect to collect after the Company has exhausted its collection procedures. The Company estimates its allowance for doubtful accounts in the period the related services are billed and reduces the allowance in future accounting periods based on write-offs during those periods. It bases the estimate for the allowance on the evaluation of historical experience of accounts going to collections and the net amounts not received. Accounts going to collection include the balances, after receipt of the approved settlements from third party payers, for the insufficient diagnosis information received from the ordering physician which result in denials of payment and our estimate of the uncollected portion of receivables from self-payers, including deductibles and copayments, which are subject to credit risk and patients’ ability to pay. The Company fully reserves through its contractual allowances amounts that have not been written off because the payer’s filing date deadline has not occurred or the collection process has not been exhausted. The Company adjusts the historical collection analysis for recoveries, if any, on an on-going basis. During fiscal 2018, the Company recorded a reclassification of approximately $1.7 million, which reduced the allowance for doubtful accounts and increased the allowance for contractual allowances; both accounts are netted against accounts receivable on the consolidated balance sheet.
The Company’s ability to collect outstanding receivables from third party payers is critical to its operating performance and cash flows. The primary collection risk lies with uninsured patients or patients for whom primary insurance has paid but a patient portion remains outstanding. The Company also assesses the current state of its billing functions in order to identify any known collection or reimbursement issues in order to assess the impact, if any, on the allowance estimates, which involves judgment. The Company believes that the collectability of its receivables is directly linked to the quality of its billing processes, most notably, those related to obtaining the accurate patient information in order to bill effectively for the services provided. Should circumstances change (e.g. shift in payer mix, decline in economic conditions or deterioration in aging of receivables), our estimates of net realizable value of receivables could be reduced by a material amount. As of July 31, 2018, approximately 23% of Clinical Labs receivables are from two payers.
Billing for laboratory services is complicated because of many factors, especially: the differences between our standard gross fee schedule for all payers and the reimbursement rates of the various payers we deal with, disparity of coverage and information requirements among the various payers, and disputes with payers as to which party is responsible for reimbursement.
The allowance for doubtful accounts as a percentage of consolidated gross accounts receivable at July 31, 2018 and 2017 was 16.9% and 19.1% respectively.
| 54 |
The following table indicates the Clinical Laboratory Services segment aged gross receivables by payer group (in thousands), which is prior to adjustment to gross receivables for: 1) contractual adjustment, 2) fully reserved balances not yet written off, and 3) other revenue adjustments.
| As of July 31, 2018 |
Total Amount |
% | Third Party Payers Amount |
% |
Medicare Amount |
% |
Self pay Amount |
% |
HMO’s Amount |
% | |||||||||||||||||||||||||
| 1-30 days | $ | 22,788 | 47 | $ | 14,886 | 48 | $ | 4,102 | 46 | $ | 864 | 15 | $ | 2,936 | 90 | ||||||||||||||||||||
| 31-60 days | 6,821 | 14 | 4,540 | 15 | 1,069 | 12 | 995 | 17 | 217 | 7 | |||||||||||||||||||||||||
| 61-90 days | 4,526 | 9 | 2,877 | 9 | 784 | 9 | 843 | 15 | 22 | 1 | |||||||||||||||||||||||||
| 91-120 days | 3,460 | 7 | 2,307 | 8 | 463 | 5 | 666 | 11 | 24 | 1 | |||||||||||||||||||||||||
| 121-150 days | 2,705 | 6 | 1,602 | 5 | 490 | 6 | 601 | 10 | 12 | — | |||||||||||||||||||||||||
| Greater than 150 days | 8,357 | 17 | 4,481 | 15 | 1,976 | 22 | 1,862 | 32 | 38 | 1 | |||||||||||||||||||||||||
| Totals | $ | 48,657 | 100 | % | $ | 30,693 | 100 | % | $ | 8,884 | 100 | % | $ | 5,831 | 100 | % | $ | 3,249 | 100 | % | |||||||||||||||
| As of July 31, 2017 |
Total Amount |
% |
Third Party Payers Amount |
% |
Medicare Amount |
% | Self pay Amount |
% |
HMO’s Amount |
% | |||||||||||||||||||||||||
| 1-30 days | $ | 25,357 | 42 | $ | 16,683 | 40 | $ | 4,022 | 60 | $ | 1,082 | 16 | $ | 3,570 | 82 | ||||||||||||||||||||
| 31-60 days | 8,732 | 15 | 5,723 | 14 | 1,294 | 19 | 1,183 | 17 | 532 | 12 | |||||||||||||||||||||||||
| 61-90 days | 5,703 | 10 | 4,208 | 10 | 529 | 8 | 927 | 14 | 39 | 1 | |||||||||||||||||||||||||
| 91-120 days | 3,749 | 6 | 2,732 | 6 | 288 | 4 | 701 | 10 | 28 | 1 | |||||||||||||||||||||||||
| 121-150 days | 3,689 | 6 | 2,772 | 7 | 228 | 3 | 672 | 10 | 17 | — | |||||||||||||||||||||||||
| Greater than 150 days | 12,455 | 21 | 9,652 | 23 | 379 | 6 | 2,270 | 33 | 154 | 4 | |||||||||||||||||||||||||
| Totals | $ | 59,685 | 100 | % | $ | 41,770 | 100 | % | $ | 6,740 | 100 | % | $ | 6,740 | 100 | % | $ | 4,340 | 100 | % | |||||||||||||||
Income Taxes
The Company accounts for income taxes under the liability method of accounting for income taxes. Under the liability method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. The liability method requires that any tax benefits recognized for net operating loss carry forwards and other items be reduced by a valuation allowance where it is not more likely than not the benefits will be realized in the foreseeable future. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. Under the liability method, the effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. It is the Company’s policy to provide for uncertain tax positions, if any, and the related interest and penalties based upon management’s assessment of whether a tax benefit is more likely than not to be sustained upon examination by tax authorities. To the extent the Company prevails in matters for which a liability for an unrecognized tax benefit is established or is required to pay amounts in excess of the liability, the Company’s effective tax rate in a given financial statement period may be affected.
Inventory
The Company values inventory at the lower of cost (first-in, first-out) or net realizable value. Work-in-process and finished goods inventories consist of material, labor, and manufacturing overhead. Write downs of inventories to net realizable value are based on a review of inventory quantities on hand and estimated sales forecasts based on sales history and anticipated future demand. Unanticipated changes in demand could have a significant impact on the value of our inventory and require additional write downs of inventory which would impact our results of operations.
Goodwill and Intangible Assets
Goodwill represents the excess of the cost of an acquisition over the fair value of the net assets acquired. Intangible assets (exclusive of patents), arose primarily from acquisitions, and primarily consist of customer relationships, trademarks, licenses, and website and database content. Finite-lived intangible assets are amortized according to their estimated useful lives, which range from 4 to 15 years. Patents represent capitalized legal costs incurred in pursuing patent applications. When such applications result in an issued patent, the related capitalized costs are amortized over a ten year period or the life of the patent, whichever is shorter, using the straight-line
| 55 |
method. The Company reviews its issued patents and pending patent applications, and if it determines to abandon a patent application or that an issued patent no longer has economic value, the unamortized balance in deferred patent costs relating to that patent is immediately expensed.
The Company tests goodwill annually as of the first day of the fourth quarter, or more frequently if indicators of potential impairment exist. In assessing goodwill for impairment, the Company has the option to first perform a qualitative assessment to determine whether the existence of events or circumstances leads to a determination that it is more likely than not that the fair value of a reporting unit is less than its carrying amount. If the Company determines that it is not more likely than not that the fair value of a reporting unit is less than its carrying amount, the Company is not required to perform any additional tests in assessing goodwill for impairment. However, if the Company concludes otherwise or elects not to perform the qualitative assessment, then it identifies the reporting units and compares the fair value of each of these reporting units to their respective carrying amount. If the carrying amount of the reporting unit is less than its fair value, no impairment exists. If the carrying amount of the reporting unit is higher than its fair value, the impairment charge is the amount by which the carrying amount exceeds its fair value, not to exceed the total amount of goodwill allocated to the reporting unit.
The Company reviews the recoverability of the carrying value of long-lived assets (including intangible assets with finite lives) of a reporting unit for impairment annually as of the first day of the fourth quarter, or more frequently if indicators of potential impairment exist. Should indicators of impairment exist, the carrying values of the assets are evaluated in relation to the operating performance and future undiscounted cash flows of the reporting unit. The net book value of the long lived asset is adjusted to fair value if its expected future undiscounted cash flow is less than its book value.
During fiscal years 2018, 2017 and 2016, there was no impairment of goodwill or long-lived assets.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
We are exposed to market risk from changes in foreign currency exchange rates resulting from activities in foreign locations (See Item 1A. Risk Factors and Note 2 in the Notes to Consolidated Financial Statements) that could impact our results of operations and financial position. We do not currently engage in any hedging or market risk management tools.
Foreign Currency Exchange Rate Risk
The financial reporting of our non-U.S. subsidiaries is denominated in currencies other than the U.S. dollar. Since the functional currency of our non-U.S. subsidiaries is the local currency, foreign currency translation adjustments are accumulated as a component of accumulated other comprehensive income in stockholders’ equity. Assuming a hypothetical decline of 10% in the exchange rates of foreign currencies against the U.S. dollar at July 31, 2018, our assets and liabilities would decrease by $0.4 million and $0.1 million, respectively, and our net sales and net earnings (loss) would decrease by $0.9 million and $0.2 million, respectively, on an annual basis.
We also maintain intercompany balances and loans receivable with subsidiaries with different local currencies. These amounts are at risk of foreign exchange losses if exchange rates fluctuate. Assuming a hypothetical increase of 10% in the exchange rates of foreign currencies against the U.S. dollar at July 31, 2018, our pre-tax earnings (loss) would be unfavorably impacted by approximately $1.4 million on an annual basis.
Interest Rate Risk
As of July 31, 2018, we have fixed interest rate financing on transportation and equipment leases.
Item 8. Financial Statements and Supplementary Data
The response to this item is submitted in a separate section of this report. See Item 15(a) (1) and (2).
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
| 56 |
Item 9A. Controls and Procedures
| 1. | Disclosure Controls and Procedures |
We maintain disclosure controls and procedures (Disclosure Controls) within the meaning of Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Our Disclosure Controls are designed to ensure that information required to be disclosed by us in the reports we file or submit under the Exchange Act, such as this Annual Report on Form 10-K, is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms. Our Disclosure Controls are also designed to ensure that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating our Disclosure Controls, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily applied its judgment in evaluating and implementing possible controls and procedures.
As of the end of the period covered by this Annual Report on Form 10-K, we evaluated the effectiveness of the design and operation of our Disclosure Controls, which was done under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer. Based on the evaluation of our Disclosure Controls, our Chief Executive Officer and Chief Financial Officer have concluded that, as of July 31, 2018, our Disclosure Controls were not effective due to material weaknesses in the Company’s internal controls over financial reporting as disclosed below under “Management’s Annual Report on Internal Control Over Financial Reporting.”
As a result of the material weaknesses identified, we performed additional analysis, substantive testing and other post-closing procedures intended to ensure our consolidated financial statements were prepared in accordance with U.S. generally accepted accounting principles. Accordingly, management believes that the consolidated financial statements and related notes included in this Annual Report on Form 10-K fairly present, in all material respects, the Company’s financial position, results of operations and cash flows for the periods presented.
| 2. | Change in Internal Control over Financial Reporting |
There were no changes in our internal control over financial reporting that occurred during the quarter ended July 31, 2018, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| 3. | Management’s Report on Internal Control Over Financial Reporting |
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, a company’s principal executive and principal financial officers and effected by the company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect of our consolidated financial statements.
There are inherent limitations on the effectiveness of any system of internal controls and procedures, including the possibility of human error and the circumvention or overriding of the controls and procedures. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies and procedures may deteriorate.
| 57 |
Accordingly, even effective internal controls and procedures can only provide reasonable assurance of achieving their control objectives.
Management assessed the effectiveness of our internal control over financial reporting as of July 31, 2018. In making this assessment, management used the criteria established in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on this assessment, management has concluded that, as of July 31, 2018, our internal control over financial reporting was not effective, as management identified deficiencies in internal control over financial reporting that were determined to be material weaknesses.
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis.
We did not design and implement effective internal control over risk assessment with regard to our processes and procedures commensurate with our financial reporting requirements, which we determined to be material weaknesses. Specifically:
| • | Our Internal Controls over Financial Reporting (“ICFR”) intended to estimate accounts receivable were insufficient to fully and timely take into account changes in the business environment and experience with ultimate collection from third-party payers in the determination of contractual adjustment amounts and collectability of accounts receivable. | |
| • | Our Information Technology General Controls (“ITGC”) intended to control change management, program access and monitoring were not adequate. These deficiencies could result in occurrences of unmonitored access to certain applications and unsupervised change management at the Information Technology level, adversely impacting the functionality of key applications, the effectiveness of process-level automated controls and the propriety of underlying data that supports the effectiveness of system generated data and reports used in financial reporting. |
The material weaknesses did not result in any material identified audit adjustments.
Our management does not expect that its disclosure controls and procedures will prevent all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur due to simple errors or mistakes. The design of any system of controls is based in part upon certain assumptions regarding the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions.
Item 5 includes the adverse audit report of EisnerAmper LLP on our internal control over financial reporting as of July 31, 2018.
| 4. | Plan to Remediate Material Weaknesses |
Management is in the process of developing a plan to remediate the material weaknesses discussed above and expects to complete the remediation of the foregoing deficiencies in the near future. Additionally, management will continue to evaluate and take actions to improve our internal control over financial reporting as changes in underlying risks affecting our business evolve. The material weaknesses will not be considered remediated until the applicable remedial controls operate for a sufficient period and management has concluded, through testing, that the controls are operating effectively.
| 5. | Report of Independent Registered Public Accounting Firm |
EisnerAmper LLP, our independent registered public accounting firm, has audited the effectiveness of the Company’s internal control over financial reporting as of July 31, 2018, as stated in their report, which is included elsewhere herein.
| 58 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
Enzo Biochem, Inc.
Opinion on the Internal Control over Financial Reporting
We have audited Enzo Biochem, Inc. and Subsidiaries’ (the “Company”) internal control over financial reporting as of July 31, 2018, based on criteria established in the Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). In our opinion, because of the effect of the material weaknesses described in the following paragraph on the achievement of the objectives of the control criteria, Enzo Biochem, Inc. and Subsidiaries has not maintained effective internal control over financial reporting as of July 31, 2018, based on criteria established in the Internal Control - Integrated Framework (2013) issued by COSO.
A material weakness is a control deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis. The following material weaknesses have been identified and included in management’s assessment: (i) The Company identified a deficiency in the operating effectiveness of a control that is considered a material weakness and (ii) several deficiencies in the operating effectiveness of other controls which in the aggregate represent a material weakness. These material weaknesses were considered in determining the nature, timing, and extent of the audit tests applied in our audit of the July 31, 2018 financial statements, and this report does not affect our report dated October 15, 2018 on those financial statements.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) ("PCAOB"), the consolidated balance sheets of Enzo Biochem, Inc. and Subsidiaries’ as of July 31, 2018, and the related consolidated statements of operations, comprehensive income (loss), stockholders’ equity, and cash flows for each of the years in the three-year period ended July 31, 2018, and the related notes and schedule, and our report dated October 15, 2018 expressed an unqualified opinion.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
An entity’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. An entity’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the entity; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the entity are being made only in accordance with authorizations of management and directors of the entity; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the entity’s assets that could have a material effect on the financial statements.
| 59 |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ EisnerAmper LLP
EISNERAMPER LLP
New York, New York
October 15, 2018
| 60 |
None
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information required under this item will be set forth in the Company’s proxy statement to be filed with the Securities and Exchange Commission on or before November 28, 2018 and is incorporated herein by reference.
Item 11. Executive Compensation
The information required under this item will be set forth in the Company’s proxy statement to be filed with the Securities and Exchange Commission on or before November 28, 2018 and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required under this item will be set forth in the Company’s proxy statement to be filed with the Securities and Exchange Commission on or before November 28, 2018 and is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required under this item will be set forth in the Company’s proxy statement to be filed with the Securities and Exchange Commission on or before November 28, 2018 and is incorporated herein by reference.
Item 14. Principal Accountant Fees and Services
The information required under this item will be set forth in the Company’s proxy statement expected to be filed with the Securities and Exchange Commission on or before November 28, 2018 and is incorporated herein by reference.
PART IV
Item 15. Exhibits, Financial Statement Schedules
| (a) | (1) | Consolidated Financial Statements | |
| Consolidated Balance Sheets - July 31, 2018 and 2017 | |||
| Consolidated Statements of Operations - Years ended July 31, 2018, 2017 and 2016 | |||
|
Consolidated Statements of Comprehensive Income (Loss) - Years ended July 31, 2018, 2017 and 2016 Consolidated Statements of Stockholders’ Equity - Years ended July 31, 2018, 2017 and 2016 |
|||
| Consolidated Statements of Cash Flows - Years ended July 31, 2018, 2017 and 2016 | |||
| Notes to Consolidated Financial Statements | |||
| (2) | Financial Statement Schedule | ||
|
Schedule II - Valuation and Qualifying Accounts |
|||
All other schedules have been omitted because the required information is included in the consolidated financial statements or the notes thereto or because they are not required. |
|||
| (3) | Exhibits |
| 61 |
The following documents are filed as Exhibits to this Annual Report on Form 10-K:
| 62 |
| 10 (v) | Purchase and Sale Agreement by and between Building Blocks Realty Co. LLC (seller) and Enzo Realty LLC (Purchaser)* | |
| 14 (a) | Code of Ethics (11) | |
| 21* | List of subsidiaries of the Company | |
| 23.1* | Consent of Independent Registered Public Accounting Firm | |
| 31 (a)* | Certification of CEO Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 31 (b)* | Certification of CFO Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 32 (a)* | Certification of CEO Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 32 (b)* | Certification of CFO Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 101. INS** | XBRL Instance Document | |
| 101. SCH** | XBRL Taxonomy Extension Schema Document | |
| 101. CAL** | XBRL Taxonomy Extension Calculation Linkbase Document | |
| 101.DEF** | XBRL Taxonomy Extension Definitions Linkbase Document | |
| 101.LAB** | XBRL Taxonomy Extension Label Linkbase Document | |
| 101.PRE** | XBRL Taxonomy Extension Presentation Linkbase Document |
| 63 |
| Notes to exhibits | ||
| * | Filed herewith | |
| ** | XBRL (Extensible Business Reporting Language) information is being furnished and not filed for purposes of Sections 11 and 12 of the Securities Act of 1933 and Section 18 of the Securities Exchange Act of 1934. | |
| (1) | The exhibits were filed as exhibits to the Company’s Registration Statement on Form S-18 (File No. 2-67359) and are incorporated herein by reference. | |
| (2) | This exhibit was filed as an exhibit to the Company’s Annual Report on Form 10-K for the year ended July 31, 1981 and is incorporated herein by reference. | |
| (3) | This exhibit was filed with the Company’s Annual Report on Form 10-K for the year ended July 31, 1989 and is incorporated herein by reference. | |
| (4) | This exhibit was filed with the Company’s Current Report on Form 8-K dated January 22, 2013 and is incorporated herein by reference. | |
| (5) | This exhibit was filed with the Company’s Annual Report on Form 10-K for the year ended July 31, 1995 and is incorporated herein by reference. | |
| (6) | This exhibit was filed with the Company’s Registration Statement on Form S-8 (333-87153) and is incorporated herein by reference. | |
| (7) | This exhibit was filed as an exhibit to the Company’s Proxy Statement of Schedule 14A filed on January 19, 2006 and is incorporated herein by reference. | |
| (8) | This exhibit was filed as appendix B to the Company’s Definitive Proxy Statement on Schedule 14A, which was filed with the Securities and Exchange Commission on November 16, 2010 and is incorporated herein by reference. | |
| (9) | This exhibit was filed with the Company’s Annual Report on Form 10-K for the year ended July 31, 2005 and is incorporated herein by reference. | |
| (10) | This exhibit was filed with the Company’s Current Report on Form 8-K on September 21, 2006 and is incorporated herein by reference. | |
| (11) | This exhibit was filed with the Company’s Annual Report on Form 10-K for the year ended July 31, 2003 and is incorporated here by reference. | |
| (12) | This exhibit was filed with the Company’s Current Report on Form 8-K on May 30, 2007 and is incorporated herein by reference. | |
| (13) | This exhibit was filed with the Company’s Current Report on Form 8-K on May 8, 2008 and is incorporated herein by reference. | |
| (14) | This exhibit was filed with the Company’s Current Report on Form 8-K on March 13, 2009 and is incorporated herein by reference. | |
| (15) | This exhibit was filed with the Company’s Current Report on Form 8-K on January 10, 2017 and is incorporated herein by reference. | |
| (16) | This exhibit was filed with the Company’s Current Report on Form 8-K on March 28, 2013 and incorporated herein by reference. | |
| (17) | This exhibit was filed with the Company’s Current Report on Form 10-K for the year ended July 31, 2013 and incorporated herein by reference. |
| 64 |
| (18) | This exhibit was filed with the Company’s Current Report on Form 8-K on April 24, 2014 and incorporated herein by reference. | |
| (19) | This exhibit was filed with the Company’s Current Report on Form 8-K on June 23, 2014 and incorporated herein by reference. | |
| (20) | This exhibit was filed with the Company’s Current Report on Form 10-K for the year ended July 31, 2014 and is incorporated herein by reference. | |
| (21) | This exhibit was filed with the Company’s Current Report on Form 8-K on July 7, 2015 and incorporated herein by reference. | |
| (22) | This exhibit was filed with the Company’s Current Report on Form 8-K on July 22, 2015 and incorporated herein by reference. | |
| (23) | This exhibit was filed with the Company’s Annual Report on Form 10-K for the year ended July 31, 2015 and is incorporated herein by reference. | |
| (24) | This exhibit was filed with the Company’s Current Report on Form 8-K on October 13, 2015 and incorporated herein by reference. | |
| (25) | This exhibit was filed with the Company’s Annual Report on Form 10-K for the year ended July 31, 2016. |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| ENZO BIOCHEM, INC. | ||||
| Date: October 15, 2018 | By: | /s/ Elazar Rabbani Ph.D. | ||
| Chairman of the Board | ||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| By: /s/ Elazar Rabbani | October 15, 2018 | ||
| Elazar Rabbani, Ph.D. | |||
| Chairman of Board of Directors and Secretary | |||
| (Principal Executive Officer) | |||
| By: /s/ Barry W. Weiner | October 15, 2018 | ||
| Barry W. Weiner, | |||
| President, Chief Financial Officer, Principal Accounting Officer, Treasurer and Director | |||
| By: /s/ Bruce A. Hanna | October 15, 2018 | ||
| Bruce A. Hanna, Ph.D., Director | |||
| By: /s/ Gregory M. Bortz | October 15, 2018 | ||
| Gregory M. Bortz, Director | |||
| By: /s/ Dov Perlysky | October 15, 2018 | ||
| Dov Perlysky, Director |
| 65 |
FORM 10-K, ITEM 15(a) (1) and (2)
ENZO BIOCHEM, INC.
LIST OF CONSOLIDATED FINANCIAL STATEMENTS
AND
FINANCIAL STATEMENT SCHEDULE
The following consolidated financial statements and financial statement schedule of Enzo Biochem, Inc. are included in Item 15(a):
All other schedules for which provision is made in the applicable accounting regulation of the Securities and Exchange Commission are not required under the related instructions or are inapplicable, and therefore have been omitted.
| F-1 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
Enzo Biochem, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Enzo Biochem, Inc. and Subsidiaries (the “Company”) as of July 31, 2018 and 2017, and the related consolidated statements of operations, comprehensive income (loss), stockholders’ equity, and cash flows for each of the years in the three-year period ended July 31, 2018, and the related notes and schedule identified in Item 15 (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the consolidated financial position of the Company as of July 31, 2018 and 2017, and the consolidated results of their operations and their cash flows for each of the years in the three-year period ended July 31, 2018, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the Company's internal control over financial reporting as of July 31, 2018, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated October 15, 2018 expressed an adverse opinion.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ EisnerAmper LLP
We have served as the Company’s auditor since 2013.
EISNERAMPER LLP
New York, New York
October 15, 2018
| F-2 |
ENZO BIOCHEM, INC.
CONSOLIDATED BALANCE SHEETS
(in thousands, except share and per share data)
| July 31, 2018 |
July 31, 2017 |
|||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 60,041 | $ | 64,167 | ||||
| Accounts receivable, net of allowance for doubtful accounts of $2,668 in 2018 and $3,576 in 2017 | 13,147 | 15,180 | ||||||
| Inventories | 7,278 | 7,047 | ||||||
| Prepaid expenses | 2,734 | 2,690 | ||||||
| Total current assets | 83,200 | 89,084 | ||||||
| Property, plant, and equipment, net | 7,636 | 7,901 | ||||||
| Goodwill | 7,452 | 7,452 | ||||||
| Intangible assets, net | 1,886 | 2,895 | ||||||
| Other | 1,486 | 333 | ||||||
| Total assets | $ | 101,660 | $ | 107,665 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable - trade | 9,516 | 10,350 | ||||||
| Accrued liabilities | 10,054 | 6,720 | ||||||
| Other current liabilities | 616 | 740 | ||||||
| Total current liabilities | 20,186 | 17,810 | ||||||
| Other liabilities | 353 | 983 | ||||||
| Total liabilities | $ | 20,539 | $ | 18,793 | ||||
| Commitments and contingencies | ||||||||
| Stockholders’ equity: | ||||||||
| Preferred Stock, $.01 par value; authorized 25,000,000 shares; no shares issued or outstanding | — | — | ||||||
| Common Stock, $.01 par value; authorized 75,000,000 shares; shares issued and outstanding: 47,182,254 at July 31, 2018 and 46,506,176 at July 31, 2017 | 472 | 465 | ||||||
| Additional paid-in capital | 330,770 | 328,294 | ||||||
| Accumulated deficit | (252,221 | ) | (241,900 | ) | ||||
| Accumulated other comprehensive income | 2,100 | 2,013 | ||||||
| Total stockholders’ equity | 81,121 | 88,872 | ||||||
| Total liabilities and stockholders’ equity | $ | 101,660 | $ | 107,665 | ||||
The accompanying notes are an integral part of these consolidated financial statements
| F-3 |
ENZO BIOCHEM, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share data)
| Years ended July 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Revenues: | ||||||||||||
| Clinical laboratory services | $ | 74,777 | $ | 77,407 | $ | 70,915 | ||||||
| Product revenues | 29,224 | 29,192 | 30,337 | |||||||||
| Royalty and license fee income | 712 | 1,205 | 1,521 | |||||||||
| Total revenues | 104,713 | 107,804 | 102,773 | |||||||||
| Operating costs, expenses and legal settlements, net: | ||||||||||||
| Cost of clinical laboratory services | 46,008 | 45,400 | 42,859 | |||||||||
| Cost of product revenues | 14,377 | 14,078 | 14,331 | |||||||||
| Research and development | 3,210 | 2,928 | 3,524 | |||||||||
| Selling, general, and administrative | 44,465 | 44,092 | 43,741 | |||||||||
| Provision for uncollectible accounts receivable | 3,690 | 2,775 | 2,336 | |||||||||
| Legal fee expense | 5,127 | 1,679 | 6,384 | |||||||||
| Legal settlements, net | — | — | (57,250 | ) | ||||||||
| Total costs, expenses and legal settlements, net | 116,877 | 110,952 | 55,925 | |||||||||
| Operating (loss) income | (12,164 | ) | (3,148 | ) | 46,848 | |||||||
| Other income (expense): | ||||||||||||
| Interest | 853 | 384 | (136 | ) | ||||||||
| Other | 168 | 125 | 122 | |||||||||
| Foreign exchange gain (loss) | (275 | ) | 135 | (474 | ) | |||||||
| (Loss) income before income taxes | (11,418 | ) | (2,504 | ) | 46,360 | |||||||
| (Provision) benefit for income taxes | 1,097 | — | (1,074 | ) | ||||||||
| Net (loss) income | $ | (10,321 | ) | $ | (2,504 | ) | $ | 45,286 | ||||
| Net (loss) income per common share: | ||||||||||||
| Basic | $ | (0.22 | ) | $ | (0.05 | ) | $ | 0.98 | ||||
| Diluted | $ | (0.22 | ) | $ | (0.05 | ) | $ | 0.97 | ||||
| Weighted average common shares outstanding: | ||||||||||||
| Basic | 46,972 | 46,350 | 46,153 | |||||||||
| Diluted | 46,972 | 46,350 | 46,602 | |||||||||
The accompanying notes are an integral part of these consolidated financial statements
| F-4 |
ENZO BIOCHEM, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
(in thousands)
| Years Ended July 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Net (loss) income | $ | (10,321 | ) | $ | (2,504 | ) | $ | 45,286 | ||||
| Other comprehensive (loss) income: | ||||||||||||
| Foreign currency translation adjustments | 87 | (186 | ) | 338 | ||||||||
| Comprehensive (loss) income | $ | (10,234 | ) | $ | (2,690 | ) | $ | 45,624 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
ENZO BIOCHEM, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
Years ended July 31, 2018, 2017, and 2016
(in thousands, except share data)
| Common Stock Shares Issued |
Treasury Stock Shares |
Common Stock Amount |
Additional Paid-in Capital |
Treasury Stock Amount |
Accumulated Deficit |
Accumulated Other Comprehensive Income |
Total Stockholders’ Equity |
|||||||||||||||||||||||||
| Balance at July 31, 2015 | 46,062,065 | — | $ | 461 | $ | 324,966 | $ | — | $ | (284,682 | ) | $ | 1,861 | $ | 42,606 | |||||||||||||||||
| Net income for the year ended July 31, 2016 | — | — | — | — | — | 45,286 | — | 45,286 | ||||||||||||||||||||||||
| Vesting of restricted stock | 11,500 | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Share-based compensation charges | — | — | — | 525 | — | — | — | 525 | ||||||||||||||||||||||||
| Issuance of common stock for employee 401(k) plan match | 160,352 | — | 2 | 707 | — | — | — | 709 | ||||||||||||||||||||||||
| Exercise of stock options | 33,702 | — | — | 90 | — | — | — | 90 | ||||||||||||||||||||||||
| Foreign currency translation adjustments | — | — | — | — | — | 338 | 338 | |||||||||||||||||||||||||
| Balance at July 31, 2016 | 46,267,619 | — | $ | 463 | $ | 326,288 | $ | — | $ | (239,396 | ) | $ | 2,199 | $ | 89,554 | |||||||||||||||||
| Net loss for the year ended July 31, 2017 | — | — | — | — | (2,504 | ) | — | (2,504 | ) | |||||||||||||||||||||||
| Vesting of restricted stock | 5,140 | — | — | — | — | — | — | |||||||||||||||||||||||||
| Share-based compensation charges | — | — | — | 831 | — | — | 831 | |||||||||||||||||||||||||
| Issuance of common stock for employee 401(k) plan match | 91,541 | — | 1 | 723 | — | — | 724 | |||||||||||||||||||||||||
| Exercise of stock options | 141,876 | — | 1 | 452 | — | — | 453 | |||||||||||||||||||||||||
| Foreign currency translation adjustments | — | — | — | — | — | — | (186 | ) | (186 | ) | ||||||||||||||||||||||
| Balance at July 31, 2017 | 46,506,176 | — | $ | 465 | $ | 328,294 | $ | — | $ | (241,900 | ) | $ | 2,013 | $ | 88,872 | |||||||||||||||||
| Net (loss) for the year ended July 31, 2018 | — | — | — | — | — | (10,321 | ) | — | (10,321 | ) | ||||||||||||||||||||||
| Vesting of restricted stock | 2,874 | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Share-based compensation charges | — | — | — | 813 | — | — | — | 813 | ||||||||||||||||||||||||
| Issuance of common stock and treasury stock for employee 401(k) plan match | 37,580 | (106,911 | ) | — | 204 | — | — | — | 204 | |||||||||||||||||||||||
| Cashless options exercise and issuance of Treasury stock | 340,898 | 106,911 | 4 | 576 | — | — | — | 580 | ||||||||||||||||||||||||
| Exercise of stock options | 294,726 | 3 | 883 | — | — | 886 | ||||||||||||||||||||||||||
| Foreign currency translation adjustments | — | — | — | — | — | — | 87 | 87 | ||||||||||||||||||||||||
| Balance at July 31, 2018 | 47,182,254 | — | $ | 472 | $ | 330,770 | $ | — | $ | (252,221 | ) | $ | 2,100 | $ | 81,121 | |||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements
| F-6 |
ENZO BIOCHEM, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| Years ended July 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Cash flows from operating activities: | ||||||||||||
| Net (loss) income | $ | (10,321 | ) | $ | (2,504 | ) | $ | 45,286 | ||||
| Adjustments to reconcile net (loss) income to net cash (used in) provided by operating activities: | ||||||||||||
| Depreciation and amortization of property, plant and equipment | 2,138 | 2,078 | 2,163 | |||||||||
| Amortization of intangible assets | 992 | 1,520 | 1,677 | |||||||||
| Provision for uncollectible accounts receivable | 3,690 | 2,775 | 2,336 | |||||||||
| Deferred income tax benefit | — | — | (60 | ) | ||||||||
| Share-based compensation charges | 813 | 831 | 525 | |||||||||
| Share-based 401(k) employer match expense | 829 | 724 | 709 | |||||||||
| Foreign exchange (gain) loss | 475 | (476 | ) | 357 | ||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Accounts receivable | (1,775 | ) | (3,244 | ) | (4,791 | ) | ||||||
| Other receivables - settlements | — | — | 6,650 | |||||||||
| Inventories | (413 | ) | 92 | 466 | ||||||||
| Prepaid expenses | (79 | ) | (618 | ) | 169 | |||||||
| Accounts payable - trade | (887 | ) | 515 | 1,103 | ||||||||
| Accrued liabilities, other current liabilities and other liabilities | 2,901 | (1,898 | ) | (3,464 | ) | |||||||
| Other assets | (1,098 | ) | — | — | ||||||||
| Total adjustments | 7,586 | 2,299 | 7,840 | |||||||||
| Net cash (used in) provided by operating activities | (2,735 | ) | (205 | ) | 53,126 | |||||||
| Cash flows from investing activities: | ||||||||||||
| Capital expenditures | (1,888 | ) | (1,753 | ) | (1,530 | ) | ||||||
| Decrease in security deposits and other | (56 | ) | 4 | 18 | ||||||||
| Net cash used in investing activities | (1,944 | ) | (1,749 | ) | (1,512 | ) | ||||||
| Cash flows from financing activities: | ||||||||||||
| Proceeds from borrowings under Credit Agreement | — | 40,694 | 89,880 | |||||||||
| Repayments under Credit Agreement | — | (42,250 | ) | (91,336 | ) | |||||||
| Installment loan payments | (327 | ) | (566 | ) | (565 | ) | ||||||
| Proceeds from exercise of stock options | 886 | 453 | 90 | |||||||||
| Net cash provided by (used in) financing activities | 559 | (1,669 | ) | (1,931 | ) | |||||||
| Effect of exchange rate changes on cash and cash equivalents | (6 | ) | 13 | (15 | ) | |||||||
| (Decrease) increase in cash and cash equivalents | (4,126 | ) | (3,610 | ) | 49,668 | |||||||
| Cash and cash equivalents - beginning of year | 64,167 | 67,777 | 18,109 | |||||||||
| Cash and cash equivalents - end of year | $ | 60,041 | $ | 64,167 | $ | 67,777 | ||||||
The accompanying notes are an integral part of these consolidated financial statements
| F-7 |
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
Note 1 - Summary of significant accounting policies
Nature of business
Enzo Biochem, Inc. (the “Company”) is an integrated life science and biotechnology company engaged in research, development, manufacturing and marketing of diagnostic and research products based on genetic engineering, biotechnology and molecular biology. These products are designed for the diagnosis of and/or screening for infectious diseases, cancers, genetic defects and other medically pertinent diagnostic information and are distributed in the United States and internationally. The Company is conducting research and development activities in the development of therapeutic products based on the Company’s technology platform of genetic modulation and immune modulation. The Company also operates a clinical laboratory that offers and provides molecular and esoteric diagnostic medical testing services in the New York, New Jersey, and Connecticut medical communities. The Company operates in three segments (see Note 15).
Principles of consolidation
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States (“U.S. GAAP”) and include the accounts of the Company and its wholly-owned subsidiaries, Enzo Clinical Labs, Inc., Enzo Life Sciences, Inc. (and its wholly-owned foreign subsidiaries), Enzo Therapeutics, Inc. and Enzo Realty LLC (“Realty”). All intercompany transactions and balances have been eliminated.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying footnotes. Actual results could differ from those estimates.
Foreign Currency Translation/Transactions
The Company has determined that the functional currency for its foreign subsidiaries is the local currency. For financial reporting purposes, assets and liabilities denominated in foreign currencies are translated at current exchange rates and profit and loss accounts are translated at weighted average exchange rates. Resulting translation gains and losses are included as a separate component of stockholders’ equity as accumulated other comprehensive income or loss. Gains or losses resulting from transactions entered into in other than the functional currency are recorded as foreign exchange gains and losses in the consolidated statements of operations.
Cash and cash equivalents
Cash and cash equivalents consist of demand deposits with banks and highly liquid money market funds. At July 31, 2018 and 2017, the Company had cash and cash equivalents in foreign bank accounts of $0.4 million and $0.5 million, respectively.
Fair Values of Financial Instruments
The recorded amounts of the Company’s cash and equivalents, receivables, loan payable, accounts payable and accrued liabilities approximate their fair values principally because of the short-term nature of these items.
Concentration of credit risk
Financial instruments that potentially subject the Company to concentrations of credit risk primarily consist of cash and cash equivalents and accounts receivable.
The Company believes the fair value of the aforementioned financial instruments approximates the cost due to the immediate or short-term nature of these items.
Concentration of credit risk with respect to the Company’s Life Sciences products segment is mitigated by the diversity of the Company’s clients and their dispersion across many different geographic regions. To reduce risk, the Company routinely assesses the financial strength of these customers and, consequently, believes that its accounts receivable credit exposure with respect to these customers is limited.
| F-8 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
The Company believes that the concentration of credit risk with respect to the Clinical Laboratory services accounts receivable is mitigated by the diversity of third party payers that insure individuals. To reduce risk, the Company routinely assesses the financial strength of these payers and, consequently, believes that its accounts receivable credit risk exposure, with respect to these payers, is limited. While the Company also has receivables due from the Federal Medicare program, the Company does not believe that these receivables represent a credit risk since the Medicare program is funded by the federal government and payment is primarily dependent on our submitting the appropriate documentation.
Accrual for Self-Funded Medical
Accruals for self-funded medical insurance are determined based on a number of assumptions and factors, including historical payment trends, claims history and current estimates. These estimated liabilities are not discounted. If actual trends differ from these estimates, the financial results could be impacted.
Revenue Recognition - Product revenues
Revenues from product sales are recognized when the products are shipped and title transfers, typically upon shipment by common carrier.
Royalties
Royalty revenues are recorded in the period earned. Royalties received in advance of being earned are recorded as deferred revenues.
Clinical laboratory services
Revenues from the Clinical Laboratory services segment are recognized upon completion of the testing process for a specific patient and reported to the ordering physician. These revenues and the associated accounts receivable are based on gross amounts billed or billable for services rendered, net of a contractual adjustment, which is the difference between amounts billed to payers and the expected reimbursable settlements from such payers.
The following table summarizes the Clinical Laboratory Services segment’s net revenues and revenue percentages by revenue category:
| Years Ended July 31, | ||||||||||||||||||||||||
| 2018 | 2017 | 2016 | ||||||||||||||||||||||
| Revenue category | Amount | % | Amount | % | Amount | % | ||||||||||||||||||
| Third-party payers | $ | 41,370 | 56 | $ | 43,059 | 56 | $ | 34,454 | 49 | |||||||||||||||
| Medicare | 12,111 | 16 | 12,705 | 16 | 11,392 | 16 | ||||||||||||||||||
| HMO’s | 11,359 | 15 | 10,263 | 13 | 10,325 | 14 | ||||||||||||||||||
| Patient self-pay | 9,937 | 13 | 11,380 | 15 | 14,744 | 21 | ||||||||||||||||||
| Total | $ | 74,777 | 100 | % | $ | 77,407 | 100 | % | $ | 70,915 | 100 | % | ||||||||||||
The Company provides services to certain patients covered by various third-party payers, including the Federal Medicare program. Laws and regulations governing Medicare are complex and subject to interpretation for which action for noncompliance includes fines, penalties and exclusion from the Medicare programs.
Other than the Medicare program, one provider whose programs are included in the “Third-party payers” and “Health Maintenance Organizations” (“HMO’s”) categories represent approximately 39%, 39% and 30% of the Clinical Laboratory Services segment net revenue for the years ended July 31, 2018, 2017 and 2016 respectively. The Company currently uses one third party reference lab for certain clinical laboratory services we provide which represents 12% of the consolidated purchases for the year ended July 31, 2018.
| F-9 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
Contractual Adjustment
The Company’s estimate of contractual adjustment is based on significant assumptions and judgments, such as its interpretation of payer reimbursement policies, and bears the risk of change. The estimation process is based on the experience of amounts approved as reimbursable and ultimately settled by payers, versus the corresponding gross amount billed to the respective payers. The contractual adjustment is an estimate that reduces gross revenue based on gross billing rates to amounts expected to be approved and reimbursed. Gross billings are based on a standard fee schedule the Company sets for all third-party payers, including Medicare, HMO’s and managed care providers. The Company adjusts the contractual adjustment estimate quarterly, based on its evaluation of current and historical settlement experience with payers, industry reimbursement trends, and other relevant factors which include the monthly and quarterly review of: 1) current gross billings and receivables and reimbursement by payer, 2) current changes in third party arrangements and 3) the growth of in-network provider arrangements and managed care plans specific to our Company.
During the years ended July 31, 2018, 2017 and 2016, the contractual adjustment percentages, determined using current and historical reimbursement statistics, were approximately 85%, 84% and 84%, respectively, of gross billings.
Accounts Receivable and Allowance for Doubtful Accounts
Accounts receivable are reported at realizable value, net of allowances for doubtful accounts, which is estimated and recorded in the period of the related revenue.
For the Clinical Laboratory Services segment, the allowance for doubtful accounts represents amounts that the Company does not expect to collect after the Company has exhausted its collection procedures. The Company estimates its allowance for doubtful accounts in the period the related services are billed and reduces the allowance in future accounting periods based on write-offs during those periods. It bases the estimate for the allowance on the evaluation of historical experience of accounts going to collections and the net amounts not received. Accounts going to collection include the balances, after receipt of the approved settlements from third party payers, for the insufficient diagnosis information received from the ordering physician which results in denials of payment, and our estimate of the uncollected portion of receivables from self-payers, including deductibles and copayments, which are subject to credit risk and patients’ ability to pay. The Company fully reserves through its contractual allowances amounts that have not been written off because the payer’s filing date deadline has not occurred or the collection process has not been exhausted. The Company adjusts the historical collection analysis for recoveries, if any, on an on-going basis. As of July 31, 2018, approximately 23% of Clinical Labs receivables are from two payers.
The Company’s ability to collect outstanding receivables from third-party payers is critical to its operating performance and cash flows. The primary collection risk lies with uninsured patients or patients for whom primary insurance has paid but a patient portion remains outstanding. The Company also assesses the current state of its billing functions in order to identify any known collection issues and to assess the impact, if any, on the allowance estimates which involves judgment. The Company believes that the collectability of its receivables is directly linked to the quality of its billing processes, most notably, those related to obtaining the correct information in order to bill effectively for the services provided. Should circumstances change (e.g. shift in payer mix, decline in economic conditions or deterioration in aging of receivables), our estimates of net realizable value of receivables could be reduced by a material amount.
The allowance for doubtful accounts as a percentage of total accounts receivable at July 31, 2018 and 2017 was 16.9% and 19.1% respectively.
The Clinical Laboratory Services segment’s net receivables are detailed by billing category and as a percent to its total net receivables. At July 31, 2018 and 2017, approximately 74% and 75% respectively, of the Company’s net accounts receivable relates to its Clinical Laboratory Services business, which operates in the New York, New Jersey and Connecticut medical communities.
The Life Sciences products segment’s accounts receivable includes royalties receivable of $0 million and $0.4 million, as of July 31, 2018 and 2017, respectively, due from QIAGEN Gaithersburg Inc. (“Qiagen”) (see Note 12).
| F-10 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
The following is a table of the Company’s net accounts receivable by segment.
| July 31, 2018 | July 31, 2017 | |||||||||||||||
| Net accounts receivable by segment | Amount | % | Amount | % | ||||||||||||
| Clinical Labs (by billing category) | ||||||||||||||||
| Third party payers | $ | 4,692 | 48 | $ | 7,256 | 64 | ||||||||||
| Patient self-pay | 2,010 | 20 | 1,591 | 14 | ||||||||||||
| Medicare | 1,740 | 18 | 1,385 | 12 | ||||||||||||
| HMO’s | 1,329 | 14 | 1,169 | 10 | ||||||||||||
| Total Clinical Labs | 9,771 | 100 | % | 11,401 | 100 | % | ||||||||||
| Total Life Sciences | 3,376 | 3,779 | ||||||||||||||
| Total accounts receivable – net | $ | 13,147 | $ | 15,180 | ||||||||||||
Changes in the Company’s allowance for doubtful accounts are as follows:
| July 31, 2018 | July 31, 2017 | |||||||
| Beginning balance | $ | 3,576 | $ | 3,517 | ||||
| Provision for doubtful accounts | 3,690 | 2,775 | ||||||
| Write-offs | (4,598 | ) | (2,716 | ) | ||||
| Ending balance | $ | 2,668 | $ | 3,576 | ||||
Inventories
The Company values inventory at the lower of cost (first-in, first-out) or net realizable value. Work-in-process and finished goods inventories consist of material, labor, and manufacturing overhead. Write downs of inventories to net realizable value are based on a review of inventory quantities on hand and estimated sales forecasts based on sales history and anticipated future demand. Unanticipated changes in demand could have a significant impact on the value of our inventory and require additional write downs of inventory which would impact our results of operations.
Property, plant and equipment
Property, plant and equipment is stated at cost, and depreciated on the straight-line basis over the estimated useful lives of the various asset classes as follows: building and building improvements: 15-30 years, and laboratory machinery and equipment and office furniture and computer equipment which range from 3-10 years. Leasehold improvements are amortized over the term of the related leases or estimated useful lives of the assets, whichever is shorter.
Goodwill and Intangible Assets
Goodwill represents the excess of the cost of an acquisition over the fair value of the net assets acquired.
Intangible assets (exclusive of patents), arose primarily from acquisitions, and primarily consist of customer relationships, trademarks, licenses, and website and database content. Finite-lived intangible assets are amortized according to their estimated useful lives, which range from 4 to 15 years. Indefinite-lived intangibles are not amortized and are evaluated each reporting period to determine whether events and circumstances continue to support their having an indefinite life. Indefinite-lived intangibles found to no longer have an indefinite life are evaluated for impairment and are then amortized over their remaining useful life as finite-lived intangible assets. Patents represent capitalized legal costs incurred in pursuing patent applications. When such applications result in an issued patent, the related capitalized costs are amortized over a ten year period or the life of the patent, whichever is shorter, using the straight-line method.
The Company reviews its issued patents and pending patent applications, and if it determines to abandon a patent application or that an issued patent no longer has economic value, the unamortized balance in deferred patent costs relating to that patent is immediately expensed.
| F-11 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
Impairment testing for Goodwill and Long-Lived Assets
The Company tests goodwill annually as of the first day of the fourth quarter, or more frequently if indicators of potential impairment exist. In assessing goodwill for impairment, the Company has the option to first perform a qualitative assessment to determine whether the existence of events or circumstances leads to a determination that it is more likely than not that the fair value of a reporting unit is less than its carrying amount. If the Company determines that it is not more likely than not that the fair value of a reporting unit is less than its carrying amount, the Company is not required to perform any additional tests in assessing goodwill for impairment. However, if the Company concludes otherwise or elects not to perform the qualitative assessment, then it identifies the reporting units and compares the fair value of each of these reporting units to their respective carrying amount. If the carrying amount of the reporting unit is less than its fair value, no impairment exists. If the carrying amount of the reporting unit is higher than its fair value, the impairment charge is the amount by which the carrying amount exceeds its fair value, not to exceed the total amount of goodwill allocated to the reporting unit. The Company performed a quantitative assessment in 2018 and 2017 and a qualitative assessment in 2016, and concluded there were no goodwill impairments.
The Company reviews the recoverability of the carrying value of long-lived assets (including intangible assets with finite lives) of a reporting unit for impairment annually as of the first day of the fourth quarter, or more frequently if indicators of potential impairment exist. Should indicators of impairment exist, the carrying values of the assets are evaluated in relation to the operating performance and future undiscounted cash flows of the reporting unit. The net book value of the long lived asset is adjusted to fair value if its expected future undiscounted cash flow is less than its book value. There were no long-lived asset impairments in 2018, 2017 or 2016.
Comprehensive income (loss)
Comprehensive income (loss) consists of the Company’s consolidated net income (loss) and foreign currency translation adjustments. Foreign currency translation adjustments included in comprehensive income (loss) were not tax effected as investments in international affiliates are deemed to be permanent. Accumulated other comprehensive income is a separate component of stockholders’ equity and consists of the cumulative foreign currency translation adjustments.
Shipping and Handling Costs
Shipping and handling costs associated with the distribution of finished goods to customers are recorded in cost of goods sold.
Research and Development
Research and development costs are charged to expense as incurred.
Advertising
All costs associated with advertising are expensed as incurred. Advertising expense, included in selling, general and administrative expense, approximated $580, $649 and $601 for the years ended July 31, 2018, 2017 and 2016, respectively.
Income Taxes
The Company accounts for income taxes under the liability method of accounting for income taxes. Under the liability method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. The liability method requires that any tax benefits recognized for net operating loss carry forwards and other items be reduced by a valuation allowance when it is more likely than not that the benefits may not be realized. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled.
Under the liability method, the effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
| F-12 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
It is the Company’s policy to provide for uncertain tax positions and the related interest and penalties based upon management’s assessment of whether a tax benefit is more likely than not to be sustained upon examination by tax authorities. At July 31, 2018, the Company believes it has appropriately accounted for any unrecognized tax benefits. To the extent the Company prevails in matters for which a liability for an unrecognized tax benefit is established or is required to pay amounts in excess of the liability, the Company’s effective tax rate in a given financial statement period may be affected.
Segment Reporting
The Company separately reports information about each operating segment that engages in business activities from which the segment may earn revenues and incur expenses, whose separate operating results are regularly reviewed by the chief operating decision maker regarding allocation of resources and performance assessment and which exceed specific quantitative thresholds related to revenue and profit or loss. The Company’s operating activities are reported in three segments (see Note 15).
Net income (loss) per share
Basic net income (loss) per share represents net income (loss) divided by the weighted average number of common shares outstanding during the period. The dilutive effect of potential common shares, consisting of outstanding stock options and unvested restricted stock, is determined using the treasury stock method. Diluted weighted average shares outstanding for fiscal 2018 and 2017 do not include the potential common shares from stock options and unvested restricted stock because to do so would have been antidilutive and as such is the same as basic weighted average shares outstanding for 2018 and 2017. For fiscal 2016, approximately 449,000 weighted average stock options were included in the calculation of diluted weighted average shares outstanding. The number of potential common shares (“in the money options”) and unvested restricted stock excluded from the calculation of diluted weighted average shares outstanding for the years ended July 31, 2018, and 2017 was 624,000, and 961,000, respectively.
For the years ended July 31, 2018, 2017 and 2016, the effect of approximately 291,000, zero and 282,000 respectively, of outstanding “out of the money” options to purchase common shares were excluded from the calculation of diluted weighted average shares outstanding because their effect would be anti-dilutive. The following table sets forth the computation of basic and diluted net loss per share for the years ended July 31:
| 2018 | 2017 | 2016 | ||||||||||
| Net (loss) income | $ | (10,321 | ) | $ | (2,504 | ) | $ | 45,286 | ||||
| Weighted-average common shares outstanding - basic | 46,972 | 46,350 | 46,153 | |||||||||
| Add: effect of dilutive stock options and restricted stock | — | — | 449 | |||||||||
| Weighted-average common shares outstanding - diluted | 46,972 | 46,350 | 46,602 | |||||||||
| Net (loss) income per share – basic | $ | (0.22 | ) | $ | (0.05 | ) | $ | 0.98 | ||||
| Net (loss) income per share – diluted | $ | (0.22 | ) | $ | (0.05 | ) | $ | 0.97 | ||||
Share-Based Compensation
The Company records compensation expense associated with stock options and restricted stock based upon the fair value of stock based awards as measured at the grant date. The Company determines the award values of stock options using the Black Scholes option pricing model.
The expense is recorded by amortizing the fair values on a straight-line basis over the vesting period, adjusted for forfeitures when they occur.
For the years ended July 31, 2018, 2017 and 2016, share-based compensation expense relating to the fair value of stock options, restricted shares and restricted stock units was approximately $813, $831, and $525, respectively (see Note 10). No excess tax benefits were recognized for the year ended July 31, 2018, 2017 and 2016.
| F-13 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
The following table sets forth the amount of expense related to share-based payment arrangements included in specific line items in the accompanying statement of operations for the years ended July 31:
| 2018 | 2017 | 2016 | ||||||||||
| Cost of clinical laboratory services | $ | — | $ | 6 | $ | 6 | ||||||
| Selling, general and administrative | 813 | 825 | 519 | |||||||||
| $ | 813 | $ | 831 | $ | 525 | |||||||
As of July 31, 2018, there was $1,138 of total unrecognized compensation cost related to non-vested share-based payment arrangements granted under the Company’s incentive stock plans, which will be recognized over a weighted average remaining life of approximately twenty four months.
Effect of New Accounting Pronouncements
Recently Adopted Accounting Pronouncements
In March 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2016-09, Improvements to Employee Share-Based Payment Accounting, which requires all excess tax benefits or deficiencies to be recognized as income tax expense or benefit in the income statement. In addition, excess tax benefits should be classified along with other income tax cash flows as an operating activity in the statement of cash flows. We adopted this standard in the fiscal year ended July 31, 2018. We recognize compensation expense by amortizing the fair values of awards on a straight basis over the vesting period, adjusted for forfeitures when they occur. The adoption of this standard did not have a material impact on our consolidated financial statements.
In May 2018, the FASB issued ASU No. 2018-05, Income Taxes (Topic 740): Amendments to SEC Paragraphs Pursuant to SEC Staff Accounting Bulletin No. 118, regarding the accounting implications of the recently issued legislation commonly referred to as the Tax Cuts and Jobs Act (the “Act”). This standard is effective immediately. The update clarifies that in a company’s financial statements that include the reporting period in which the Act was enacted, the company must first reflect the income tax effects of the Act in which the accounting under U.S. GAAP is complete. These amounts would not be provisional amounts. The company would also report provisional amounts for those specific income tax effects for which the accounting under U.S. GAAP is incomplete but a reasonable estimate can be determined. We have recorded a provisional amount which we believe is a reasonable estimate of the effects of the Act on our financial statements as of July 31, 2018. Technical corrections or other forthcoming guidance could change how we interpret provisions of the Act, which may impact our effective tax rate and could affect our deferred tax assets, tax positions and/or our tax liabilities.
Pronouncements Issued but Not Yet Adopted
In May 2014, FASB issued ASU No. 2014-09, Revenue from Contracts with Customers: Topic 606. This ASU and its amendments supersede existing revenue recognition guidance, including industry-specific guidance. The core principle of the revenue recognition standard is to require an entity to recognize as revenue the amount that reflects the consideration which it expects to be entitled to in exchange for the goods or services it transfers control of to its customers.
We will adopt this ASU in the first quarter of our fiscal year beginning August 1, 2018 using the full retrospective method. We continue to assess the impact of this ASU on our results of operations, financial position, cash flows and disclosures. Based on our assessment of this ASU, the majority of the amounts that were historically classified as the provision for uncollectible accounts receivable, primarily related to patient responsibility, will be considered an implicit price concession in determining revenues from clinical laboratory services. Accordingly, we will report uncollectible balances associated with patient responsibility as a reduction of the transaction price and therefore as a reduction in revenues from clinical laboratory services, when historically these amounts were classified as the provision for uncollectible accounts receivable within operating costs and expenses. The residual balance of the provision for uncollectible accounts receivable will also be reclassified and included in selling, general and administrative expense. As a result of the adoption of this ASU, we preliminarily estimate the following impact to our consolidated statements of operations for the years ended July 31, 2018 and 2017:
| F-14 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
| Year Ended July 31, 2018 | Year Ended July 31, 2017 | ||||||||
| As Reported |
Adjustment for ASU on Revenue Recognition |
Reclassification of Residual |
As Adjusted |
As Reported |
Adjustment for ASU on Revenue Recognition |
Reclassification of Residual |
As Adjusted |
||
| Total Revenues | $104,7133 | $(3,700) | - | $101,013 | $107,804 | $(2,718) | - | $105,086 | |
| Provision for uncollectible accounts receivable | 3,690 | (3,700) | $10 | - | 2,775 | (2,718) | $(57) | - | |
| Selling, general and administrative expenses | 44,435 | - | (10) | 44,425 | 44,092 | - | 57 | 44,149 | |
| Net loss | $(10,321) | - | - | $(10,321) | $(2,504) | - | - | $(2,504) | |
In addition, the adoption of this ASU will result in increased disclosure, including qualitative and quantitative disclosures about the nature, amount, timing and uncertainty of revenue and cash flows arising from contracts with customers. However, the adoption of this ASU is not expected to have a material impact on our financial position or cash flows.
In February 2016, FASB issued ASU No. 2016-02 – Leases (Topic 842), as amended. The new standard establishes a right-of-use (ROU) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the income statement. The new standard is effective for our fiscal year beginning August 1, 2019 including interim periods within that fiscal year. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. As amended in July 2018, an additional and optional transition method to adopt the new leases standard was established. Under this new transition method, an entity initially applies the new leases standard at the adoption date and recognizes a cumulative-effect adjustment to the opening balance of retained earnings in the period of adoption. Consequently, an entity’s reporting for the comparative periods presented in the financial statements in which it adopts the new leases standard will continue to be in accordance with current GAAP (Topic 840, Leases).
We believe the adoption of this standard would materially impact our consolidated financial statements by significantly increasing our non-current assets and non-current liabilities on our consolidated balance sheets if we record the right of use assets and related lease liabilities for our existing operating leases. We will recognize expense in the consolidated statement of operations similar to current lease accounting, in the cost of sales and selling, general and administrative.
In June 2016, FASB issued ASU No. 2016-13 Financial Instruments – Credit Losses (Topic 326). This standard changes the impairment model for most financial instruments, including trade receivables, from an incurred loss method to a new forward-looking approach, based on expected losses. The estimate of expected credit losses will require entities to incorporate considerations of
| F-15 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
historical information, current information and reasonable and supportable forecasts. Adoption of this standard is required for our annual and interim periods beginning August 1, 2020 and must be adopted using a modified retrospective transition approach. We are currently assessing the impact of the adoption of this standard on our results of operations, financial position and cash flows.
In May 2017, the FASB issued ASU 2017-09, Compensation – Stock Compensation (Topic 718) Scope of Modification Accounting which provides guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. Adoption of this standard is required for our annual and interim periods beginning August 1, 2018 with the amendments in the update applied prospectively to an award modified on or after the adoption date. Based on our preliminary assessment of the standard, we expect that any excess income tax benefits or deficiencies from stock-based compensation, which would be recognized as discrete items within income tax expense rather than additional paid in capital, would be offset by an equivalent adjustment to the deferred tax valuation allowance. Accordingly, we expect that the adoption of this standard will have no impact on our reported operations for the foreseeable future. In addition, we expect to continue to account for award forfeitures in the period they occur.
We reviewed all other recently issued accounting pronouncements and have concluded they are not applicable or not expected to be significant to the accounting for our operations.
Reclassification
Certain prior period amounts have been reclassified to conform to the current period presentation. These reclassifications had no effect on the reported results of operations.
Note 2 - Goodwill and intangible assets
Goodwill
The Company’s net carrying amount of goodwill is in the Clinical Laboratory Services segment and is $7,452 as of July 31, 2018 and 2017.
Intangible assets
The Company’s change in the net carrying amount of intangible assets, all in the Life Sciences Products segment is as follows:
| Gross | Accumulated Amortization |
Net | ||||||||||
| July 31, 2016 | $ | 27,650 | $ | (23,228 | ) | $ | 4,422 | |||||
| Amortization expense | — | (1,520 | ) | (1,520 | ) | |||||||
| Foreign currency translation | (214 | ) | 207 | (7 | ) | |||||||
| July 31, 2017 | $ | 27,436 | $ | (24,541 | ) | $ | 2,895 | |||||
| Amortization expense | — | (992 | ) | (992 | ) | |||||||
| Foreign currency translation | (89 | ) | 72 | (17 | ) | |||||||
| July 31, 2018 | $ | 27,347 | $ | (25,461 | ) | $ | 1,886 | |||||
| F-16 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
Intangible assets, all finite-lived, consist of the following:
| July 31, 2018 | July 31, 2017 | ||||||||||||||||||||||
| Gross | Accumulated Amortization |
Net | Gross | Accumulated Amortization |
Net | ||||||||||||||||||
| Patents | $ | 11,027 | $ | (10,980 | ) | $ | 47 | $ | 11,027 | $ | (10,951 | ) | $ | 76 | |||||||||
| Customer relationships | 11,836 | (9,997 | ) | 1,839 | 11,881 | (9,083 | ) | 2,798 | |||||||||||||||
| Website and acquired content | 1,008 | (1,008 | ) | — | 1,011 | (1,011 | ) | — | |||||||||||||||
| Licensed technology and other | 483 | (483 | ) | — | 484 | (463 | ) | 21 | |||||||||||||||
| Trademarks | 2,993 | (2,993 | ) | — | 3,033 | (3,033 | ) | — | |||||||||||||||
| Total | $ | 27,347 | $ | (25,461 | ) | $ | 1,886 | $ | 27,436 | $ | (24,541 | ) | $ | 2,895 | |||||||||
At July 31, 2018 information with respect to the intangibles acquired is as follows:
| Useful life assigned |
Weighted average remaining useful life |
|||
| Customer relationships | 8-15 years | 2 years | ||
| Other intangibles | 10 years | 4 years |
At July 31, 2018, the weighted average remaining useful life of intangible assets was approximately two years.
Estimated amortization expense related to these finite-lived intangible assets for the five succeeding fiscal years ending July 31 is as follows:
| 2019 | $ | 873 | ||
| 2020 | 508 | |||
| 2021 | 272 | |||
| 2022 | 233 | |||
| 2023 | — |
Amortization expense for the years ended July 31, 2018, 2017, and 2016 was $992, $1,520, and $1,677, respectively.
Note 3 - Supplemental disclosure for statement of cash flows
In the years ended July 31, 2018, 2017, and 2016, income taxes paid by the Company approximated $65, $1,021, and $279 respectively.
In the years ended July 31, 2018, 2017, and 2016, interest paid by the Company approximated $76, $119, and $121 respectively.
During fiscal 2018, 2017 and 2016, the Company financed $0, $69 and $95, respectively, in machinery and transportation equipment under installment loans.
During fiscal 2018 and 2017, the Company did not enter into any capital lease agreements. During fiscal 2016, the Company entered into capital lease agreements for machinery and equipment with a cost basis of $1,280.
Note 4 - Inventories
Inventories consisted of the following at July 31:
| 2018 | 2017 | |||||||
| Raw materials | $ | 754 | $ | 852 | ||||
| Work in process | 2,174 | 1,905 | ||||||
| Finished products | 4,350 | 4,290 | ||||||
| $ | 7,278 | $ | 7,047 | |||||
| F-17 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
Note 5 - Property, plant, and equipment
At July 31, 2018 and 2017 property, plant, and equipment consist of:
| 2018 | 2017 | |||||||
| Building and building improvements | $ | 4,917 | $ | 4,898 | ||||
| Machinery and equipment (includes asset under capital lease - see Note 9) | 7,570 | 7,878 | ||||||
| Office furniture and computer equipment | 15,362 | 19,434 | ||||||
| Leasehold improvements | 5,262 | 5,389 | ||||||
| 33,111 | 37,599 | |||||||
| Accumulated depreciation and amortization | (26,187 | ) | (30,410 | ) | ||||
| 6,924 | 7,189 | |||||||
| Land and land improvements | 712 | 712 | ||||||
| $ | 7,636 | $ | 7,901 | |||||
Note 6 - Income taxes
The benefit (provision) for income taxes for fiscal years ended July 31 is as follows:
| 2018 | 2017 | 2016 | ||||||||||
| Federal | $ | 1,097 | $ | — | $ | (968 | ) | |||||
| State and local | — | — | (121 | ) | ||||||||
| Foreign | — | — | (45 | ) | ||||||||
| Deferred benefit | — | — | 60 | |||||||||
| Benefit (provision) for income taxes | $ | 1,097 | $ | — | $ | (1,074 | ) | |||||
On December 22, 2017, the Act was signed into law making significant changes to the Internal Revenue Code. Changes include, but are not limited to, a corporate tax rate decrease from 35% to 21% effective for tax years beginning after December 31, 2017, the transition of U.S. international taxation from a worldwide tax system to a territorial system, and a one-time transition tax on the mandatory deemed repatriation of cumulative foreign earnings as of December 31, 2017.
The Act also puts in place new tax laws that will apply prospectively, which include, but are not limited to, (1) implementing a base erosion and anti-abuse tax, (2) generally eliminating U.S. federal income taxes on dividends from foreign subsidiaries, (3) a new provision designed to tax currently in the U.S. global intangible low-taxed income (“GILTI”) of foreign subsidiaries, which allows for the possibility of utilizing foreign tax credits to offset the income tax liability (subject to some limitations), and (4) a lower effective U.S. tax rate on certain revenues from sources outside the U.S.
The Company calculated its best estimate of the impact of the Act in accordance with its understanding of the Act and guidance available as of the date of this filing and recorded a $1.1 million tax benefit in the 2018 period in which the legislation was enacted, related to a credit for alternative minimum taxes (AMT) paid in prior periods. A provisional amount related the remeasurement of certain deferred tax assets and liabilities based on the rates at which they are expected to reverse in the future resulted in a charge of $11.5 million which was fully offset by an equivalent adjustment to the deferred tax valuation allowance. No provisional amount related to the one-time transition tax on the mandatory deemed repatriation of foreign earnings was deemed necessary.
On December 22, 2017, Staff Accounting Bulletin No. 118 (“SAB 118”) was issued to address the application of U.S. GAAP in situations when a registrant does not have the necessary information available, prepared or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the Act. In accordance with SAB 118, the Company has determined that the $1.1 million benefit recorded which relates to the AMT credit is a provisional amount and a reasonable estimate as of July 31, 2018.
Deferred tax assets and liabilities arise from temporary differences between the tax basis of assets and liabilities and their reported amounts in the financial statements. The components of deferred tax assets (liabilities) as of July 31 are as follows:
| F-18 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
| 2018 | 2017 | |||||||
| Deferred tax assets: | ||||||||
| Federal tax carryforward losses | $ | 13,975 | $ | 18,831 | ||||
| Provision for uncollectible accounts receivable | 791 | 1,392 | ||||||
| State and local tax carry forward losses | 352 | — | ||||||
| Accrued royalties | 102 | 149 | ||||||
| Stock compensation | 575 | 782 | ||||||
| Depreciation | 581 | 804 | ||||||
| Research and development and other tax credit carryforwards | 1,286 | 2,208 | ||||||
| Foreign tax carryforward losses | 2,771 | 2,420 | ||||||
| Intangibles | 1,811 | 2,888 | ||||||
| Inventory | 1,786 | 2,536 | ||||||
| Accrued expenses | 1,194 | 1,420 | ||||||
| Other, net | 58 | 69 | ||||||
| Deferred tax assets | 25,282 | 33,499 | ||||||
| Prepaid expenses | (772 | ) | (863 | ) | ||||
| Other, net | (39 | ) | (55 | ) | ||||
| Deferred tax liabilities | (811 | ) | (918 | ) | ||||
| Net deferred tax assets before valuation allowance | 24,471 | 32,581 | ||||||
| Less: valuation allowance | (24,471 | ) | (32,581 | ) | ||||
| Net deferred tax liabilities | $ | — | $ | — | ||||
The Company recorded a valuation allowance during the years ended July 31, 2018 and 2017 equal to domestic and certain foreign net deferred tax assets. The Company believes that the valuation allowance is necessary as it is not more likely than not that the deferred tax assets will be realized in the foreseeable future based on positive and negative evidence available at this time. This conclusion was reached because of uncertainties relating to future taxable income, in terms of both its timing and its sufficiency, which would enable the Company to realize the deferred tax assets. For fiscal year 2018 and 2017 the change in the valuation allowance was $8.1 million and $2.3 million, respectively.
As of July 31, 2018, the Company had U.S. federal net operating loss carryforwards of approximately $65.8 million. The U.S. federal tax loss carryforwards, if not fully utilized, expire between 2030 and 2038. Utilization is dependent on generating sufficient taxable income prior to expiration of the tax loss carryforwards. In addition, the Company has research and development tax credit carryforwards of approximately $1.3 million which expire between 2025 and 2038. As of July 31, 2018, the Company has state net operating loss carryforwards of approximately $6.5 million, which if not fully utilized, expire between 2037 and 2038. As of July 31, 2018, the Company had foreign loss carryforwards of approximately $10.8 million.
The components of income (loss) before income taxes consisted of the following for the years ended July 31:
| 2018 | 2017 | 2016 | ||||||||||
| United States operations | $ | (9,540 | ) | $ | (212 | ) | $ | 48,692 | ||||
| International operations | (1,878 | ) | (2,292 | ) | (2,332 | ) | ||||||
| Income (loss) before taxes | $ | (11,418 | ) | $ | (2,504 | ) | $ | 46,360 | ||||
The benefit (provision) for income taxes was at rates different from U.S. federal statutory rates for the following reasons for the years ended July 31:
| F-19 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
| 2018 | 2017 | 2016 | ||||||||||
| Federal statutory rate | 26.4 | % | 34.0 | % | (34.0 | )% | ||||||
| Penalties and other expenses not deductible for income tax return purposes | (1.0 | ) | (14.5 | ) | (1.1 | ) | ||||||
| State income taxes, net of benefit of federal tax deduction | — | — | (0.1 | ) | ||||||||
| Change in valuation allowance | 73.9 | 64.0 | 32.9 | |||||||||
| State tax law change | — | (81.1 | ) | — | ||||||||
| Impact of Tax Act on valuation allowance | (100.1 | ) | — | — | ||||||||
| AMT refund under Tax Act | 9.6 | — | — | |||||||||
| Other | 0.8 | (2.4 | ) | — | ||||||||
| 9.6 | % | — | % | (2.3 | )% | |||||||
Because there are no undistributed earnings at the Company’s foreign subsidiaries at July 31, 2018, no U.S. federal income taxes have been provided. As of July 31, 2018, the Company has no liabilities for uncertain tax positions. It is the Company’s policy to record interest and penalties as a component of tax expense. The Company files income tax returns in the U.S. Federal jurisdiction, various U.S. state jurisdictions and several foreign jurisdictions.
Note 7 - Loan Payable
In June 2013, the Company entered into a secured Revolving Loan and Security Agreement (the “Credit Agreement”) among the Company and certain of its subsidiaries, with Enzo Therapeutics as a guarantor, and MidCap Financial LLC. (formerly Healthcare Finance Group, LLC). The Credit Agreement, which expired in December 2016, provided for borrowings against eligible US receivables, as defined, of the Clinical Laboratory Services and Life Science Products segments up to $8.0 million at defined eligibility percentages and provided for additional borrowings of $4.0 million for increased eligible assets. Debt issuance costs of $281 were amortized over the life of the Credit Agreement. The balance of unamortized debt issuance cost was zero and $28 at July 31, 2017 and 2016, respectively, and was included in prepaid expenses. The nominal interest rate for the four month period the loan was outstanding during fiscal year 2017 and year ended July 31, 2016 was 5.25%. The effective interest rate for the credit agreement was 14.3% for the four month period the loan was outstanding in fiscal 2017 and 11.4% for the fiscal year ended July 31, 2016. The Credit Agreement expired and was repaid in full on December 7, 2016.
Note 8 - Accrued Liabilities
At July 31 accrued liabilities consist of:
| 2018 | 2017 | |||||||
| Payroll, benefits, severance and commissions | $ | 4,870 | $ | 4,092 | ||||
| Legal | 2,121 | 442 | ||||||
| Professional fees | 811 | 599 | ||||||
| Research and development | — | 143 | ||||||
| Other | 2,252 | 1,444 | ||||||
| $ | 10,054 | $ | 6,720 | |||||
Self-Insured Medical Plan
The Company self-funds medical insurance coverage for certain of its U.S. based employees. The risk to the Company is believed to be limited through the use of individual and aggregate stop loss insurance. As of July 31, 2018 and 2017, the Company has established a reserve of $0.3 million, respectively, which is included in accrued liabilities, for claims that have been reported but not paid and incurred but not reported. The reserve is based upon the Company’s historical payment trends, claim history and current estimates.
Note 9 - Other liabilities
At July 31 Other liabilities consist of:
| 2018 | 2017 | |||||||
| Settlement | $ | — | $ | 410 | ||||
| Capital lease obligation | 351 | 551 | ||||||
| Installment loans | 2 | 22 | ||||||
| $ | 353 | $ | 983 | |||||
| F-20 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
The capital lease obligation and installment loans are for machinery and equipment used in the Clinical Laboratory Services segment. Amortization of the assets recorded under the capital lease is included in depreciation expense. At July 31, 2018, the accumulated amortization on the capital lease was $782 and the imputed interest rate ranges from 2.99% to 9.5%.
| Future minimum lease and loan payments are as follows: | Capital lease | Installment loans |
||||||
| 2019 | $ | 173 | $ | 24 | ||||
| 2020 | 244 | 2 | ||||||
| 2021 | 126 | — | ||||||
| 2022 | — | — | ||||||
| Total payments | 543 | 26 | ||||||
| Less: interest | (19 | ) | (1 | ) | ||||
| Total net of interest | 524 | 25 | ||||||
| Less: current portion | (173 | ) | (23 | ) | ||||
| Other liabilities - net | $ | 351 | $ | 2 | ||||
The weighted average interest rate on our short term borrowings during fiscal 2018 and 2017 was 7.2% and 7.5%, respectively.
Note 10 - Stockholders’ equity
Controlled Equity Offering
The Company has a Controlled Equity OfferingSM Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co., as sales agent (“Cantor”). Under the Sales Agreement, the Company may offer and sell, from time to time, through Cantor, shares of the Company’s common stock, par value $0.01 per share (the “Common Stock”). The Company pays Cantor a commission of 3.0% of the aggregate gross proceeds received under the Sale Agreement. The Company is not obligated to make any sales of the Shares under the Sales Agreement. The offering of Shares pursuant to the Sales Agreement will terminate upon the earlier of (a) the sale of all of the Shares subject to the Sales Agreement or (b) the termination of the Sales Agreement by Cantor or the Company, as permitted therein. The initial agreement contemplated the sale of shares of the Company’s common stock having an aggregate offering price of up to $20.0 million. In December 2014, the Sales Agreement was amended in order for the Company to offer and sell additional shares of Common Stock having an aggregate offering price of $20.0 million.
On September 1, 2017, the Company filed with the SEC a “shelf” registration and sales agreement prospectus covering the offering, issuance and sale of our Common Stock that may be issued and sold under the existing Sales Agreement in an aggregate amount of up to $19.2 million. A total of $150 million of securities may be sold under this shelf registration, which was declared effective September 15, 2017.
For the years ended July 31, 2018 and 2017, the Company did not sell any shares of common stock under the Sales Agreement.
Treasury stock
During fiscal year 2018, certain officers of the Company exercised 340,898 stock options in non-cash transactions. The officers surrendered 106,911 previously acquired shares of the Company’s common stock to exercise the stock options. The Company recorded approximately $1,014, the market value of the surrendered shares, as treasury stock. All of the treasury shares were subsequently reissued in the share-based 401(k) employer match made during fiscal year ended July 31, 2018.
Common stock
In fiscal 2018, the Company issued 106,911 shares of treasury stock and 37,580 shares of common stock for its employees’ 401(k) matching contributions obligation. The Company recorded an expense of $782 for the match, representing the fair value of the shares at the date of issuance.
In fiscal 2017, the Company issued 91,541 shares of common stock for its employees’ 401(k) matching contributions obligation. The Company recorded an expense of $724 for the match representing the fair value of the shares at the date of issuance.
| F-21 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
In fiscal 2016, the Company issued 160,352 shares of common stock for its employees’ 401(k) matching contributions obligation. The Company recorded an expense of $709 for the match representing the fair value of the shares at the date of issuance.
Incentive stock plans
On January 14, 2011, the Company’s stockholders approved the adoption of the 2011 Incentive Plan (the “2011 Plan”) which provides for the issuance of equity awards, including among others, options, restricted stock and restricted stock units for up to 3,000,000 Common Shares. The exercise price of options granted under the 2011 Plan, and consistent with other Plans, is equal to or greater than fair market value of the Common Stock on the date of grant. Unless terminated earlier by the Board of Directors, the 2011 Plan will terminate at the earliest of; (a) such time as no shares of Common Stock remain available for issuance under the 2011 Plan or (b) tenth anniversary of the effective date of the 2011 Plan. On January 5, 2018, the Company’s stockholders approved the amendment and restatement of the 2011 Plan to increase the number of shares available for issuance by 2,000,000 bringing the total number of shares available for award under the 2011 Plan to 5,000,000. Awards outstanding upon expiration of the 2011 Plan shall remain in effect until they have been exercised, terminated, or have expired. As of July 31, 2018, there were approximately 1,930,200 shares available for grant under the 2011 Plan.
The Company estimates the fair value of each stock option award on the measurement date using a Black-Scholes option pricing model. The fair value of awards is amortized to expense on a straight line basis over the requisite service period. The Company expenses restricted stock awards based on vesting requirements, primarily time elapsed.
Options granted pursuant to the plans may be either incentive stock options or non-statutory options The 2011 Plan provides for the issuance of stock options, restricted stock and restricted stock unit awards which generally vest over a two to four year period. A summary of the activity pursuant to the Company’s stock option plans for the years ended July 31, 2017, 2016, and 2015 is as follows:
| 2018 | 2017 | 2016 | ||||||||||||||||||||||
| Options | Weighted - Average Exercise Price |
Options | Weighted - Average Exercise Price |
Options | Weighted - Average Exercise Price |
|||||||||||||||||||
| Outstanding at beginning of year | 2,132,995 | $ | 4.26 | 1,813,875 | $ | 3.43 | 1,358,104 | $ | 3.04 | |||||||||||||||
| New Grants | 415,580 | $ | 5.57 | 493,996 | $ | 7.07 | 495,473 | $ | 4.48 | |||||||||||||||
| Exercised | (635,625 | ) | $ | 2.98 | (141,876 | ) | $ | 3.19 | (33,702 | ) | $ | 2.81 | ||||||||||||
| Expired | (30,834 | ) | $ | 5.97 | (33,000 | ) | $ | 5.29 | (6,000 | ) | $ | 3.61 | ||||||||||||
| Outstanding at end of year | 1,882,116 | $ | 4.99 | 2,132,995 | $ | 4.26 | 1,813,875 | $ | 3.43 | |||||||||||||||
| Exercisable at end of year | 1,149,489 | $ | 4.28 | 1,388,475 | $ | 3.27 | 1,094,794 | $ | 2.95 | |||||||||||||||
| Weighted average fair value of options granted during year |
$ | 1.91 | $ | 2.45 | $ | 1.56 | ||||||||||||||||||
The intrinsic value of stock option awards that vested during the fiscal year represents the value of the Company’s closing stock price on the last trading day of the fiscal year in excess of the exercise price multiplied by the number of options that vested. Total intrinsic value of options that vested and were exercisable during the fiscal years ended July 31, 2018, 2017, and 2016 was $743, $10,530 and $4,399, respectively. The intrinsic value of options outstanding at July 31, 2018, 2017, and 2016 was $1,723, $14,510 and $6,451, respectively. The intrinsic value of the options exercised in fiscal 2018, 2017 and 2016 was $6,014, $1,388 and $64, respectively.
| F-22 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
Listed below are the assumptions used to fair value options granted during fiscal years 2018, 2017 and 2016:
| Grant Date |
Options Granted |
Exercise Price |
Term (years) |
Vesting Period (years) |
FMV of options Granted/Per Share |
Expected Life (years) |
Expected Volatility % |
Interest Rate % |
Vested Shares at 7/31/2018 |
|||||||||
| 7/31/18 | 170,000 | $ 4.42 | 5 | 2 | $ 1.53 | 3.25 | 44.88 | 2.78 | — | |||||||||
| 7/31/18 | 115,000 | $ 4.42 | 5 | 3 | $ 1.61 | 3.50 | 45.92 | 2.79 | — | |||||||||
| 7/5/18 | 10,000 | $ 5.52 | 5 | 3 | $ 2.02 | 3.50 | 46.14 | 2.67 | — | |||||||||
| 5/31/18 | 5,000 | $ 6.50 | 5 | 3 | $ 2.34 | 3.50 | 45.52 | 2.09 | — | |||||||||
| 1/5/18 | 110,580 | $ 8.36 | 5 | 2 | $ 2.71 | 3.25 | 42.85 | 2.09 | — | |||||||||
| 1/2/18 | 5,000 | $ 8.25 | 5 | 3 | $ 2.76 | 3.50 | 42.59 | 2.07 | — | |||||||||
| 1/5/17 | 264,896 | $ 7.07 | 5 | 2 | $ 2.40 | 3.25 | 46.28 | 1.48 | 132,448 | |||||||||
| 1/5/17 | 229,100 | $ 7.07 | 5 | 3 | $ 2.48 | 3.50 | 45.85 | 1.54 | 66,533 | |||||||||
| 3/14/16 | 112,000 | $ 4.35 | 5 | 2 | $ 1.46 | 3.25 | 46.13 | 1.19 | 112,000 | |||||||||
| 3/14/16 | 191,600 | $ 4.35 | 5 | 3 | $ 1.57 | 3.50 | 48.14 | 1.24 | 77,865 | |||||||||
| 6/16/16 | 2,000 | $ 5.61 | 5 | 2 | $ 1.88 | 3.25 | 46.64 | 0.85 | 2,000 | |||||||||
| 1/6/16 | 189,873 | $ 4.66 | 5 | 2 | $ 1.65 | 3.25 | 48.60 | 1.31 | 189,873 |
The following table summarizes information for stock options outstanding at July 31, 2018:
| Options outstanding and exercisable | ||||||||||||
| Range of Exercise prices | Shares | Weighted-Average Remaining Contractual Life in Years |
Weighted-Average Exercise Price |
|||||||||
| $2.53 - $4.41 | 790,168 | 0.63 | $ | 3.48 | ||||||||
| $4.42 - $7.07 | 976,368 | 1.15 | $ | 5.75 | ||||||||
| $7.08 - $8.36 | 115,580 | 0.27 | $ | 8.36 | ||||||||
| 1,882,116 | ||||||||||||
Restricted Stock Awards
During fiscal 2017, the compensation committee of the Company’s board of directors approved grants of restricted stock and restricted stock unit awards (the “Awards”) to certain officers and certain employees under the 2011 Plan. The Awards vest upon the recipient’s continued employment service rateably over four years. Share-based compensation expense is based on the fair value of the award as measured on the grant date and is recorded over the vesting period on a straight-line basis. The Awards will be forfeited if the recipient ceases to be employed by the Company, as defined in the Plans’ terms. The Awards settle in shares of the Company’s common stock on a one-for-one basis.
| F-23 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
The following table summarizes the activity pursuant to restricted stock awards for the years ended July 31,
| 2018 | 2017 | 2016 | ||||||||||||||||||||||
| Awards | Weighted - Average Grant Date Fair Value |
Awards | Weighted - Average Grant Date Fair Value |
Awards | Weighted - Average Grant Date Fair Value |
|||||||||||||||||||
| Outstanding at beginning of year | 7,436 | $ | 4.45 | 8,501 | $ | 4.13 | 21,501 | $ | 8.84 | |||||||||||||||
| Awarded | — | $ | — | 4,250 | $ | 6.95 | — | $ | — | |||||||||||||||
| Vested | (2,874 | ) | $ | (5.20 | ) | (5,140 | ) | $ | (4.10 | ) | (11,500 | ) | $ | (2.83 | ) | |||||||||
| Forfeited | (1,949 | ) | $ | (5.18 | ) | (175 | ) | $ | (2.14 | ) | (1,500 | ) | $ | (2.86 | ) | |||||||||
| Outstanding (non-vested) at end of year | 2,613 | $ | 1.74 | 7,436 | $ | 4.45 | 8,501 | $ | 4.13 | |||||||||||||||
| Weighted average market value of awards granted during year | $ | — | $ | 6.95 | $ | — | ||||||||||||||||||
The fair value of the awards that vested during the years ended July 31, 2018, 2017 and 2016 was $24, $44 and $46, respectively.
Performance Stock Units
To better align the long-term interest of executives with growing U.S. practices, beginning in fiscal 2018, the Company granted long-term incentive awards in the form of time based stock options and performance-based restricted stock units (“Performance Stock Units” or “PSUs”). The PSUs earned will be determined over a three-year performance period. The primary performance metrics will be revenue and Adjusted EBITDA growth. Payouts based on revenue and adjusted EBITDA goals will be modified based on Total Shareholder Return (“TSR”) performance relative to Enzo’s peer group.
On July 31, 2018, the Company awarded a total of 32,000 PSUs to its executive officers, this award provides for the grant of shares of our common stock at the end of a three –year period based on the achievement of average revenue growth and adjusted EBITDA growth over that period. For fiscal 2018 the Company did not accrue any compensation expense for these PSU’s as the three-year performance period has just begun and achievement of the growth goals is currently not probable. At July 31, 2018, the fair value of this award was $141.
Note 11 - Employee benefit plan
The Company has a qualified Salary Reduction Profit Sharing Plan (the “Plan”) for eligible U.S. employees under Section 401(k) of the Internal Revenue Code. The Plan provides for voluntary employee contributions through salary reduction and voluntary employer contributions at the discretion of the Company. For the years ended July 31, 2018, 2017, and 2016, the Company authorized employer matched contributions of 50% of the employees’ contribution up to 10% of the employees’ compensation, payable in Enzo Biochem, Inc. common stock. The share-based 401(k) employer matched contribution was approximately $781, $724, and $709 in fiscal years 2018, 2017, and 2016, respectively. As of July 31, 2018, 2017 and 2016 the Company accrued a total of $493, $412 and $413 in 401(K) matching contributions within the Accrued liabilities account.
The Company’s Swiss operations provide a pension plan named the Enzo Life Sciences (ELS) AG Vertrag - Nr. 2/401144, (the “Swiss Plan”) under the Swiss government’s social security system for Swiss employees. The current required minimum saving contribution is 13% for employees over age 25 and minimum annual investment return is 1.00%. Employees are required to contribute based on a formula and the Company’s Swiss operations make contributions of at least 50% of the employee contribution. The status of the Swiss Plan, which is substantially funded as of December 31, 2017, the latest plan year end, is as follows:
| As of December 31, | 2017 | 2016 | 2015 | |||||||||
| Total Assets | $ | 1,849 | $ | 2,184 | $ | 2,149 | ||||||
| Accumulated Benefit Obligation | $ | 2,094 | $ | 2,432 | $ | 2,397 | ||||||
| Funded status | 88 | % | 90 | % | 90 | % | ||||||
| Fiscal Year ended July 31, | 2017 | 2016 | 2015 | |||||||||
| Contributions | $ | 205 | $ | 224 | $ | 224 | ||||||
| F-24 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
The Swiss Plan’s contract expires December 31, 2019 and currently the Company has no plans to change the current funding or plan design. No events have occurred that would impact the Swiss Plan status.
Note 12 - Royalty and other income
The Company had a license agreement with Qiagen that began in 2005, whereby the Company earned quarterly running royalties on the net sales of Qiagen products subject to a license until the underlying patent expired on April 24, 2018. During the years ended July 31, 2018, 2017 and 2016, the Company recorded royalty income under the agreement of approximately $712, $1,205 and $1,521 respectively, which is included in the Life Sciences products segment.
Note 13 - Commitments
Leases
The Company leases equipment, office and laboratory space under several non-cancellable operating leases that expire through June 2028. Certain leases include renewal options and rent escalation clauses. An entity owned by certain executive officers/directors of the Company owns the building that the Company leases as its main facility for clinical laboratory operations and certain research operations. In addition to the minimum annual rentals of space, the lease is subject to annual increases, based on the consumer price index. Annual increases are limited to 3% per year. Rent expense for this lease, inclusive of real estate taxes, approximated $1,798, $1,752, and $1,704 (net of real estate tax abatement received of $61 for 2016) during fiscal years 2018, 2017 and 2016, respectively. Total rent expense incurred by the Company during fiscal 2018, 2017 and 2016 for all its facilities was approximately $4,398, $4,658 and $4,572, respectively.
Minimum future annual rentals under all non-cancellable operating leases, net of sublease rental income of $323, as of July 31, 2018, are as follows:
| Years ended July 31, | ||||||
| 2019 | $ | 6,809 | ||||
| 2020 | 5,091 | |||||
| 2021 | 3,553 | |||||
| 2022 | 3,018 | |||||
| 2023 | 2,858 | |||||
| Thereafter | 12,295 | |||||
| $ | 33,626 | |||||
Employment Agreements
The Company has employment agreements with certain officers that are cancellable at any time but provide for severance pay in the event an officer is terminated by the Company without cause, as defined in the agreements. Unless cancelled earlier or with notice as defined, the agreement automatically renews for two years. Aggregate minimum compensation commitments, exclusive of any severance provisions as of July 31, 2018 is $2,498.
Note 14 - Contingencies
As of July 31, 2018, there are seven cases that are either pending or on appeal, which were originally brought by the Company in the United States District Court for the District of Delaware (“the Court”), alleging patent infringement against various companies. On June 28, 2017, the Court issued an opinion in the Gen-Probe case, granting Gen-Probe’s motion for summary judgment that the asserted claims of the ’180 patent are invalid for nonenablement. The Court entered final judgment of invalidity of the asserted claims of the ‘180 patent on July 19, 2017 in the Gen-Probe and Hologic cases. The Court entered partial final judgment of invalidity of the asserted claims of the ‘180 patent and stayed the remainder of the cases in the Becton Dickinson and Roche cases on July 31, 2017 and August 2, 2017, respectively. The Company filed notices of appeal in each of the Gen-Probe, Hologic, Becton Dickinson, and Roche cases, which were docketed by the United States Court of Appeals for the Federal Circuit (“Federal Circuit”). In the Abbott case, the parties agreed that the Court’s summary judgment ruling in the Gen-Probe case invalidated all of the ’180 patent claims
| F-25 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
asserted against the Abbott Defendants. On August 15, 2017, the Court granted Abbott’s motion for summary judgment that the asserted claims of the ’405 patent are invalid for nonenablement. On September 1, 2017, the Court entered final judgment of invalidity of the asserted claims of the ‘180 and ‘405 patents for nonenablement in the Abbott case. Enzo subsequently filed a notice of appeal in the Abbott case on September 14, 2017. The Federal Circuit docketed the appeal on September 15, 2017. The Federal Circuit consolidated the appeals from the Abbott, Becton Dickinson, Gen-Probe, Hologic, and Roche litigations (“Consolidated Appeals”). We disagree with the Court’s invalidity decisions regarding the ‘180 and ‘405 patents in the pending cases as set forth in our opening brief in the Consolidated Appeals pending in the Federal Circuit filed on November 28, 2017. In the Consolidated Appeals, we have asked the Federal Circuit to reverse the Court’s grants of final and summary judgment of invalidity of the asserted claims of the ‘180 and ‘405 patents and to remand the cases against Abbott, Becton Dickinson, Gen-Probe, Hologic, and Roche to the Court. Briefing is now complete in the Consolidated Appeals. The parties await the Federal Circuit’s scheduling of an oral argument date for the Consolidated Appeals. In the other two cases involving Hologic, one of the cases is stayed (Hologic II), while the other case (Hologic III) that involves the ‘581 patent is proceeding under the Court’s scheduling order. In Hologic III, the Court granted Enzo’s motion to amend its complaint to add two new defendants, Grifols Diagnostic Solutions, Inc. and Grifols, S.A, to that case. The parties have completed claim construction briefing and a claim construction hearing, but the Court has not issued a claim construction order. The Court amended the scheduling order such that fact discovery will close on October 31, 2019, dispositive motions will be heard on May 7, 2019, and trial will begin on November 18, 2019. Regarding Hologic’s petition requesting institution of an inter partes review proceeding of U.S. Patent No. 6,221,581 (“the ‘581 patent”) filed with the United States Patent and Trademark Office (“PTO”), the Patent Trial and Appeals Board (“the Board”) denied institution of Hologic’s petition on April 18, 2018. On May 18, 2018, Hologic filed with the Board, a request for rehearing of the order denying institution of inter partes review of the ‘581 patent. Enzo filed a brief in response to Hologic’s request for rehearing.
As of July 31, 2018, the Company and Enzo Life Sciences are engaged in litigation in the United States District Court for the Southern District of New York against Roche Diagnostic GmbH and its related company Roche Molecular Systems, Inc. (“Roche”), as declaratory judgment defendants. This case was commenced in May 2004. Roche seeks a declaratory judgment of non-breach of contract and patent invalidity against the Company and Enzo Life Sciences. Roche has also asserted tort claims against the Company and Enzo Life Sciences. The Company and Enzo Life Sciences have asserted breach of contract and patent infringement causes of action against Roche. There has been extensive discovery. In 2011, Roche moved for summary judgment of non-infringement regarding the Company’s patent claims. In 2012, the motion was granted in part and denied in part. In December 2012, Roche moved for summary judgment on the Company’s non-patent claims. Additional discovery was taken and the Company responded to the motions in May 2013. In December 2013, the Court granted in part and denied in part Roche’s summary judgment motion. In October 2014, the Court ordered that damages discovery concerning the Company’s remaining contract and patent claims and Roche’s claims should be completed by the end of January 2015, and expert discovery should be completed following the Court’s claim construction ruling concerning the Company’s patent infringement claim against Roche. Roche dropped its tort claims during damages discovery. On October 2, 2017, the Court issued its claim construction ruling. On September 8, 2018, the Court issued an order (i) directing that motions for summary judgment should be filed on October 10, 2018 and a proposed pretrial order by February 22, 2019, and (ii) scheduling an April 8, 2019 trial. The Company and Enzo Life Sciences intend to vigorously press their remaining claims and contest the claims against them.
The following legal settlements are included in the statement of operations under Legal settlements, net within the Life Science segment for the 2016 period:
The Company and the U.S. Department of Justice reached a settlement agreement to resolve an investigation focused primarily on an alleged failure to collect diagnosis codes from physicians who ordered tests through Enzo Clinical Labs, and recorded a charge of $2.0 million during fiscal year 2014. The settlement amount is being paid with interest over a five-year period. During fiscal year 2016, the Company accrued an additional $1.5 million, in the statement of operations under legal settlements, net within the Clinical Labs segment, due to the Company’s achievement of certain financial milestones. As of July 31, 2018, the total liability for this settlement is $0.4 million and is included in other current liabilities.
In June 2014, the Company, as plaintiff finalized and executed a settlement agreement with PerkinElmer, Inc., and PerkinElmer Health Sciences, Inc. (together, “PerkinElmer”). PerkinElmer paid $7.0 million in escrow pursuant to the agreement because of a former attorney’s charging lien for fees allegedly owed for past services rendered to the Company. In December 2015, the Company entered into a Settlement Agreement with the former attorney pursuant to which the Company and the former attorney resolved their respective claims against each other. In January 2016, the Company received a total of approximately $7.0 million from the escrow referred to above in accordance with the terms of the Settlement Agreement. In October 2015, the Company reached and finalized a settlement with Affymetrix, Inc. in the amount of $6.8 million, net in a patent infringement action brought by the Company.
| F-26 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
In January 2016, the Company reached and finalized a settlement agreement with Agilent Technologies, Inc. in the amount of $6.1 million, net in a patent infringement action brought by the Company. In May 2016, the Company reached and finalized a settlement with Life Technologies Corporation in the amount of $24.3 million, net in an infringement action brought by the Company. In July 2016, the Company reached and finalized a settlement with Illumina, Inc., in the amount of $14.5 million, net in an infringement action brought by the Company.
There can be no assurance that the Company will be successful in these litigations. Even if the Company is not successful, management does not believe that there will be a significant adverse monetary impact on the Company.
The Company is party to other claims, legal actions, complaints, and contractual disputes that arise in the ordinary course of business. The Company believes that any liability that may ultimately result from the resolution of these matters will not, individually or in the aggregate, have a material adverse effect on its financial position or results of operations
Note 15 - Segment reporting
The Company has three reportable segments: Life Sciences Products, Clinical Laboratory Services and Therapeutics. The Company’s Life Sciences Products segment develops, manufactures, and markets products to research and pharmaceutical customers. The Clinical Laboratory Services segment provides diagnostic services to the health care community. The Company’s Therapeutics segment conducts research and development activities for therapeutic drug candidates. The Company evaluates segment performance based on segment income (loss) before taxes. Costs excluded from segment income (loss) before taxes and reported as “Other” consist of corporate general and administrative costs which are not allocable to the three reportable segments.
Legal fee expense incurred to defend the Company’s intellectual property, which may result in settlements recognized in another segment and other general corporate matters are considered a component of the Other segment. Legal fee expense specific to other segments’ activities have been allocated to those segments.
Legal settlements, net, represent activities for which royalties would have been received in the Company’s Life Sciences Products segment and expenses related to an investigation within the Clinical Laboratory Services segment. Management of the Company assesses assets on a consolidated basis only and therefore, assets by reportable segment have not been included in the reportable segments below. The accounting policies of the reportable segments are the same as those described in the summary of significant accounting policies.
| F-27 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
The following financial information represents the operating results of the reportable segments of the Company:
Year ended July 31, 2018
| Clinical Laboratory Services |
Life Sciences Products |
Therapeutics | Other | Consolidated | ||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Clinical laboratory services | $ | 74,777 | — | — | — | $ | 74,777 | |||||||||||||
| Product revenues | — | $ | 29,224 | — | — | 29,224 | ||||||||||||||
| Royalty and license fee income | — | 712 | — | — | 712 | |||||||||||||||
| Total revenues | 74,777 | 29,936 | — | — | 104,713 | |||||||||||||||
| Operating costs and expenses: | ||||||||||||||||||||
| Cost of clinical laboratory services | 46,008 | — | — | — | 46,008 | |||||||||||||||
| Cost of product revenues | — | 14,377 | — | — | 14,377 | |||||||||||||||
| Research and development | — | 2,305 | $ | 905 | — | 3,210 | ||||||||||||||
| Selling, general and administrative | 24,656 | 11,627 | — | $ | 8,182 | 44,465 | ||||||||||||||
| Provision for uncollectible accounts receivable | 3,700 | (10 | ) | — | — | 3,690 | ||||||||||||||
| Legal fee expense | 67 | 58 | — | 5,002 | 5,127 | |||||||||||||||
| Total operating costs and expenses | 74,431 | 28,357 | 905 | 13,184 | 116,877 | |||||||||||||||
| Operating income (loss) | 346 | 1,579 | (905 | ) | (13,184 | ) | (12,164 | ) | ||||||||||||
| Other income (expense) | ||||||||||||||||||||
| Interest | (91 | ) | 50 | — | 894 | 853 | ||||||||||||||
| Other | 29 | 11 | — | 128 | 168 | |||||||||||||||
| Foreign exchange gain | — | (275 | ) | — | — | (275 | ) | |||||||||||||
| Income (loss) before taxes | $ | 284 | $ | 1,365 | $ | (905 | ) | $ | (12,162 | ) | $ | (11,418 | ) | |||||||
| Depreciation and amortization included above | $ | 1,667 | $ | 1,387 | $ | — | $ | 76 | $ | 3,130 | ||||||||||
| Share-based compensation included in above: | ||||||||||||||||||||
| Selling, general and administrative | 125 | 79 | — | $ | 609 | 813 | ||||||||||||||
| Total | $ | 125 | $ | 79 | $ | — | $ | 609 | $ | 813 | ||||||||||
| Capital expenditures | $ | 1,685 | $ | 203 | $ | — | $ | — | $ | 1,888 | ||||||||||
| F-28 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
The following financial information represents the operating results of the reportable segments of the Company:
Year ended July 31, 2017
| Clinical Laboratory Services |
Life Sciences Products |
Therapeutics | Other | Consolidated | ||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Clinical laboratory services | $ | 77,407 | — | — | — | $ | 77,407 | |||||||||||||
| Product revenues | — | $ | 29,192 | — | — | 29,192 | ||||||||||||||
| Royalty and license fee income | — | 1,205 | — | — | 1,205 | |||||||||||||||
| Total revenues | 77,407 | 30,397 | — | — | 107,804 | |||||||||||||||
| Operating costs, expenses and legal settlements, net: | ||||||||||||||||||||
| Cost of clinical laboratory services | 45,400 | — | — | — | 45,400 | |||||||||||||||
| Cost of product revenues | — | 14,078 | — | — | 14,078 | |||||||||||||||
| Research and development | — | 2,311 | $ | 617 | — | 2,928 | ||||||||||||||
| Selling, general and administrative | 24,465 | 11,232 | — | $ | 8,395 | 44,092 | ||||||||||||||
| Provision for uncollectible accounts receivable | 2,718 | 57 | — | — | 2,775 | |||||||||||||||
| Legal fee expense | 146 | 79 | — | 1,454 | 1,679 | |||||||||||||||
| Total operating costs, expenses and legal settlements, net | 72,729 | 27,757 | 617 | 9,767 | 110,952 | |||||||||||||||
| Operating income (loss) | 4,678 | 2,640 | (617 | ) | (9,767 | ) | (3,148 | ) | ||||||||||||
| Other income (expense) | ||||||||||||||||||||
| Interest | (112 | ) | 46 | — | 450 | 384 | ||||||||||||||
| Other | 137 | (60 | ) | — | 48 | 125 | ||||||||||||||
| Foreign exchange gain | — | 135 | — | — | 135 | |||||||||||||||
| Income (loss) before taxes | $ | 4,703 | $ | 2,761 | $ | (617 | ) | $ | (9,269 | ) | $ | (2,504 | ) | |||||||
| Depreciation and amortization included above | $ | 1,586 | $ | 1,913 | $ | — | $ | 99 | $ | 3,598 | ||||||||||
| Share-based compensation included in above: | ||||||||||||||||||||
| Cost of clinical laboratory services | $ | 6 | $ | — | $ | — | $ | — | $ | 6 | ||||||||||
| Selling, general and administrative | 111 | 74 | — | $ | 640 | 825 | ||||||||||||||
| Total | $ | 117 | $ | 74 | $ | — | $ | 640 | $ | 831 | ||||||||||
| Capital expenditures | $ | 1,363 | $ | 390 | $ | — | $ | — | $ | 1,753 | ||||||||||
| F-29 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
The following financial information represents the operating results of the reportable segments of the Company:
Year ended July 31, 2016
|
Clinical Laboratory Services |
Life Sciences Products |
Therapeutics | Other | Consolidated | ||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Clinical laboratory services | $ | 70,915 | — | — | — | $ | 70,915 | |||||||||||||
| Product revenues | — | $ | 30,337 | — | — | 30,337 | ||||||||||||||
| Royalty and license fee income | — | 1,521 | — | — | 1,521 | |||||||||||||||
| Total revenues | 70,915 | 31,858 | — | — | 102,773 | |||||||||||||||
| Operating costs, expenses and legal settlements, net: | ||||||||||||||||||||
| Cost of clinical laboratory services | 42,859 | — | — | — | 42,859 | |||||||||||||||
| Cost of product revenues | — | 14,331 | — | — | 14,331 | |||||||||||||||
| Research and development | — | 2,720 | $ | 804 | — | 3,524 | ||||||||||||||
| Selling, general and administrative | 22,882 | 11,761 | — | $ | 9,098 | 43,741 | ||||||||||||||
| Provision for uncollectible accounts receivable | 2,375 | (39 | ) | — | — | 2,336 | ||||||||||||||
| Legal fee expense | 134 | 11 | — | 6,239 | 6,384 | |||||||||||||||
| Legal settlements, net | 1,500 | (58,750 | ) | — | — | (57,250 | ) | |||||||||||||
| Total operating costs, expenses and legal settlements, net | 69,750 | (29,966 | ) | 804 | 15,337 | 55,925 | ||||||||||||||
| Operating (loss) income | 1,165 | 61,824 | (804 | ) | (15,337 | ) | 46,848 | |||||||||||||
| Other income (expense) | ||||||||||||||||||||
| Interest | (105 | ) | 47 | — | (78 | ) | (136 | ) | ||||||||||||
| Other | 10 | 38 | — | 74 | 122 | |||||||||||||||
| Foreign exchange gain | — | (474 | ) | — | — | (474 | ) | |||||||||||||
| (Loss) income before income taxes | $ | 1,070 | $ | 61,435 | $ | (804 | ) | $ | (15,341 | ) | $ | 46,360 | ||||||||
| Depreciation and amortization included above | $ | 1,676 | $ | 2,091 | $ | — | $ | 73 | $ | 3,840 | ||||||||||
| Share-based compensation included in above: | ||||||||||||||||||||
| Cost of clinical laboratory services | $ | 6 | $ | — | $ | — | $ | — | $ | 6 | ||||||||||
| Selling, general and administrative | 51 | 30 | — | $ | 438 | 519 | ||||||||||||||
| Total | $ | 57 | $ | 30 | $ | — | $ | 438 | $ | 525 | ||||||||||
| Capital expenditures | $ | 1,216 | $ | 314 | $ | — | $ | — | $ | 1,530 | ||||||||||
| F-30 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
Geographic financial information is as follows:
| Net sales to unaffiliated customers: | 2018 | 2017 | 2016 | |||||||||
| United States | $ | 95,388 | $ | 99,469 | $ | 94,016 | ||||||
| Switzerland | 2,584 | 2,371 | 2,709 | |||||||||
| United Kingdom | 1,851 | 1,673 | 1,730 | |||||||||
| Other international countries | 4,890 | 4,291 | 4,318 | |||||||||
| Total | $ | 104,713 | $ | 107,804 | $ | 102,773 | ||||||
| Long-lived assets at July 31, | 2018 | 2017 | ||||||||||
| United States | $ | 16,210 | $ | 17,241 | ||||||||
| Switzerland | 502 | 649 | ||||||||||
| United Kingdom | 127 | 185 | ||||||||||
| Other international countries | 135 | 173 | ||||||||||
| Total | $ | 16,974 | $ | 18,248 | ||||||||
The Company’s reportable segments are determined based on the services they perform, the products they sell, and the royalties and license fee income they earn, not on the geographic area in which they operate. The Company’s Clinical Laboratory Services segment operates 100% in the United States with all revenue derived there. The Life Sciences Products segment earns product revenue both in the United States and foreign countries and royalty and license fee income in the United States. The following is a summary of the Life Sciences Products segment revenues attributable to customers located in the United States and foreign countries:
| 2018 | 2017 | 2016 | ||||||||||
| United States | $ | 19,898 | $ | 22,062 | $ | 23,102 | ||||||
| Foreign countries | 9,326 | 8,335 | 8,756 | |||||||||
| $ | 29,224 | $ | 30,397 | $ | 31,858 | |||||||
Note 16 - Summary of Selected Quarterly Financial Data (unaudited)
The following table contains statement of operations information for each quarter of the years ended July 31, 2018 and 2017. The Company believes that the following information reflects all normal recurring adjustments necessary for a fair presentation of the information for the periods presented. The operating results for any quarter are not necessarily indicative of results for any future period.
Unaudited quarterly financial data for fiscal 2018 and 2017 is summarized as follows:
| Quarter Ended | ||||||||||||||||
| Fiscal 2018 |
October 31, 2017 |
January 31, 2018 |
April 30, 2018 |
July 31, 2018 |
||||||||||||
| Total revenues | $ | 27,676 | $ | 26,952 | $ | 25,630 | $ | 24,455 | ||||||||
| Gross profit | 12,245 | 11,345 | 11,073 | 9,665 | ||||||||||||
| Income (loss) before income taxes | (640 | ) | (1,998 | ) | (3,016 | ) | (5,764 | ) | ||||||||
| Net loss | (640 | ) | (901 | ) | (3,016 | ) | (5,764 | ) | ||||||||
| Basic and diluted loss per common share | $ | (0.01 | ) | $ | (0.02 | ) | $ | (0.06 | ) | $ | (0.12 | ) | ||||
| Quarter Ended | ||||||||||||||||
| Fiscal 2017 |
October 31, 2016 |
January 31, 2017 |
April 30, 2017 |
July 31, 2017 |
||||||||||||
| Total revenues | $ | 26,284 | $ | 26,260 | $ | 27,089 | $ | 28,171 | ||||||||
| Gross profit | 12,079 | 11,688 | 12,173 | 12,386 | ||||||||||||
| Income (loss) before income taxes | (1,454 | ) | (1,000 | ) | (39 | ) | 71 | |||||||||
| Net income (loss) | (1,474 | ) | (1,053 | ) | (71 | ) | 94 | |||||||||
| Basic and diluted loss per common share | $ | (0.03 | ) | $ | (0.02 | ) | $ | (0.00 | ) | $ | 0.00 | |||||
| F-31 |
ENZO BIOCHEM, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 31, 2018
(Dollars in thousands except share data)
Note 17 – Subsequent Event
On August 27, 2018, the Company announced that as part of implementing its growth strategy, it has entered into an agreement to purchase a commercial facility with nearly 36,000 square feet in Farmingdale, NY at a price of approximately $6.0 million. The purchased of this facility extends Enzo’s New York campus to nearly 101,000 square feet, complementing the Company’s existing sites in Michigan, Switzerland, France and Belgium.
| F-32 |
ENZO BIOCHEM,
INC
SCHEDULE II
VALUATION AND QUALIFYING ACCOUNTS
Years ended July 31, 2018, 2017 and 2016
(in thousands)
|
Year ended July 31, |
Description |
Balance at Beginning of period |
Charged (credited) to costs and expenses |
Charged to other accounts |
Deductions |
Balance at end of period |
|||||||||||||
| 2018 | Allowance for doubtful accounts receivable | 3,576 | 3,690 | 4,598 | (1) | 2,668 | |||||||||||||
| 2017 | Allowance for doubtful accounts receivable | 3,517 | 2,775 | 2,716 | (1) | 3,576 | |||||||||||||
| 2016 | Allowance for doubtful accounts receivable | 1,786 | 2,336 | 605 | (1) | 3,517 | |||||||||||||
| 2018 | Deferred tax valuation allowance | 32,581 | (8,110 | ) | 24,471 | ||||||||||||||
| 2017 | Deferred tax valuation allowance | 34,912 | (2,331 | ) | 32,581 | ||||||||||||||
| 2016 | Deferred tax valuation allowance | 49,658 | (14,746 | ) | 34,912 | ||||||||||||||
| (1) | Write-off of uncollectible accounts receivable. |
| S-1 |